

Diagnosis and treatment of hyperinsulinaemic hypoglycaemia and its implications for paediatric endocrinology
- PMID: 28855921
- PMCID: PMC5575922
- DOI: 10.1186/s13633-017-0048-8
Diagnosis and treatment of hyperinsulinaemic hypoglycaemia and its implications for paediatric endocrinology
Abstract
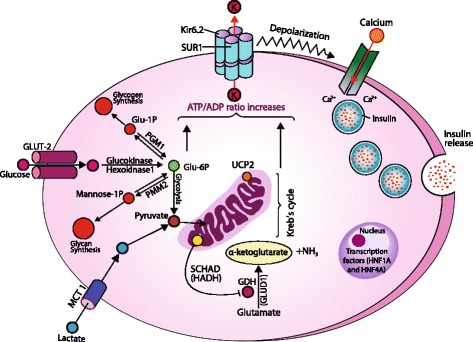
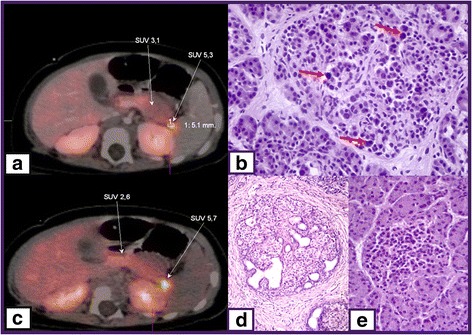
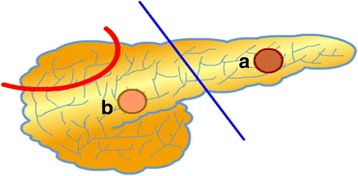
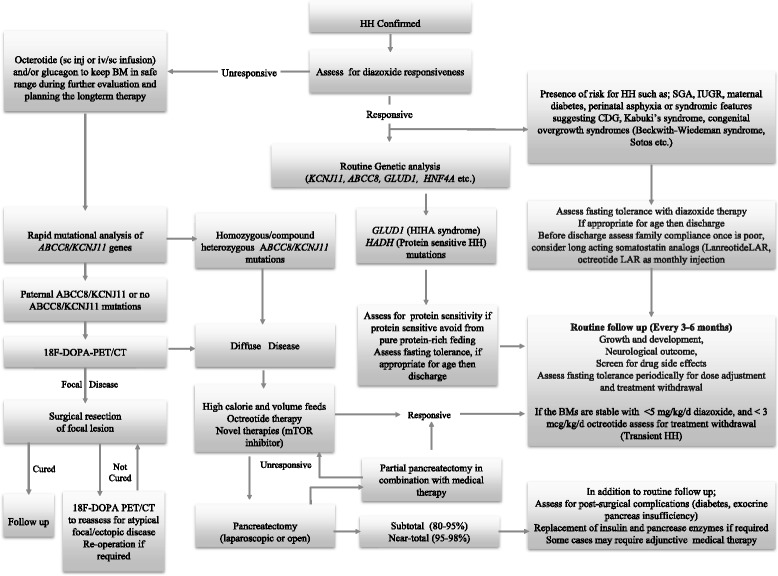
Glucose homeostasis requires appropriate and synchronous coordination of metabolic events and hormonal activities to keep plasma glucose concentrations in a narrow range of 3.5-5.5 mmol/L. Insulin, the only glucose lowering hormone secreted from pancreatic β-cells, plays the key role in glucose homeostasis. Insulin release from pancreatic β-cells is mainly regulated by intracellular ATP-generating metabolic pathways. Hyperinsulinaemic hypoglycaemia (HH), the most common cause of severe and persistent hypoglycaemia in neonates and children, is the inappropriate secretion of insulin which occurs despite low plasma glucose levels leading to severe and persistent hypoketotic hypoglycaemia. Mutations in 12 different key genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, UCP2, HNF4A, HNF1A, HK1, PGM1 and PMM2) constitute the underlying molecular mechanisms of congenital HH. Since insulin supressess ketogenesis, the alternative energy source to the brain, a prompt diagnosis and immediate management of HH is essential to avoid irreversible hypoglycaemic brain damage in children. Advances in molecular genetics, imaging methods (18F-DOPA PET-CT), medical therapy and surgical approach (laparoscopic and open pancreatectomy) have changed the management and improved the outcome of patients with HH. This up to date review article provides a background to the diagnosis, molecular genetics, recent advances and therapeutic options in the field of HH in children.
Keywords: Children; Congenital hyperinsulinaemia; Diffuse; Focal; Hyperinsulinaemic hypoglycaemia; Sirolimus.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Shah P, Rahman SA, Demirbilek H, Güemes M, Hussain K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol 2016. doi: 10.1016/S2213-8587(16)30323-0. - PubMed
-
- Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol. 2003;149(1):43–51. - PubMed
-
- Arya VB, Flanagan SE, Kumaran A, Shield JP, Ellard S, Hussain K, Kapoor RR. Clinical and molecular characterisation of hyperinsulinaemic hypoglycaemia in infants born small-for-gestational age. Arch Dis Child Fetal Neonatal Ed. 2013;98:F356–F358. doi: 10.1136/archdischild-2012-302880. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
