

β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson's disease
- PMID: 28860381
- PMCID: PMC5761666
- DOI: 10.1126/science.aaf3934
β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson's disease
Abstract
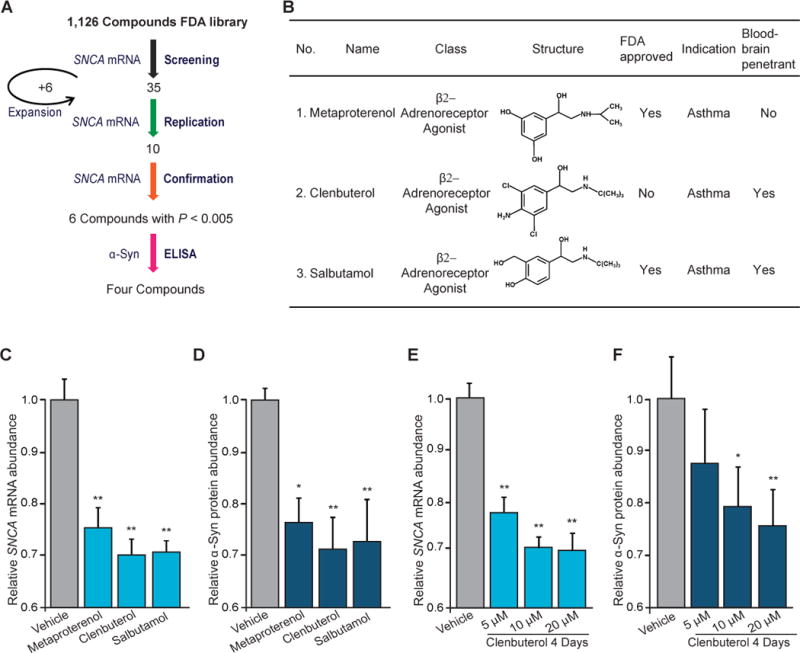
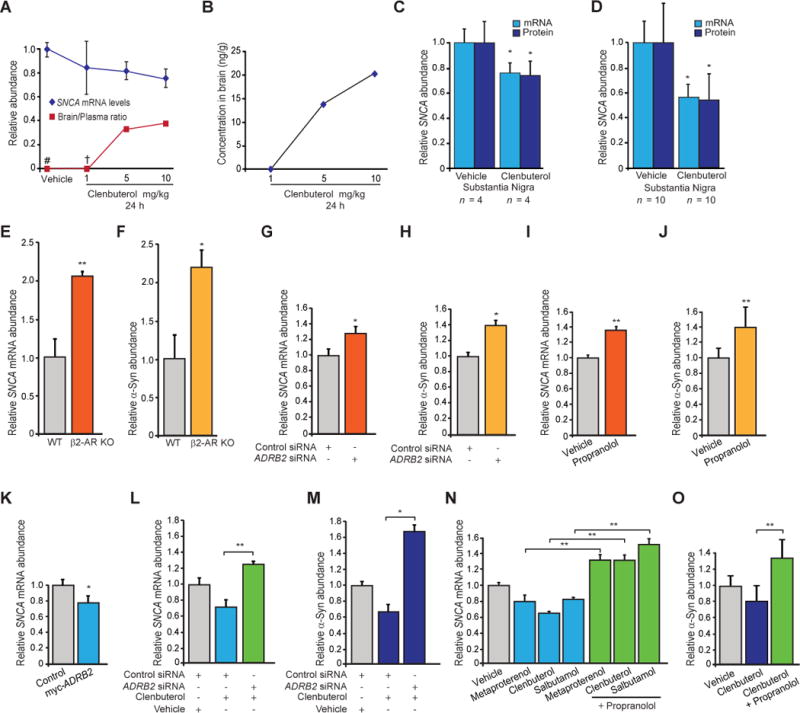
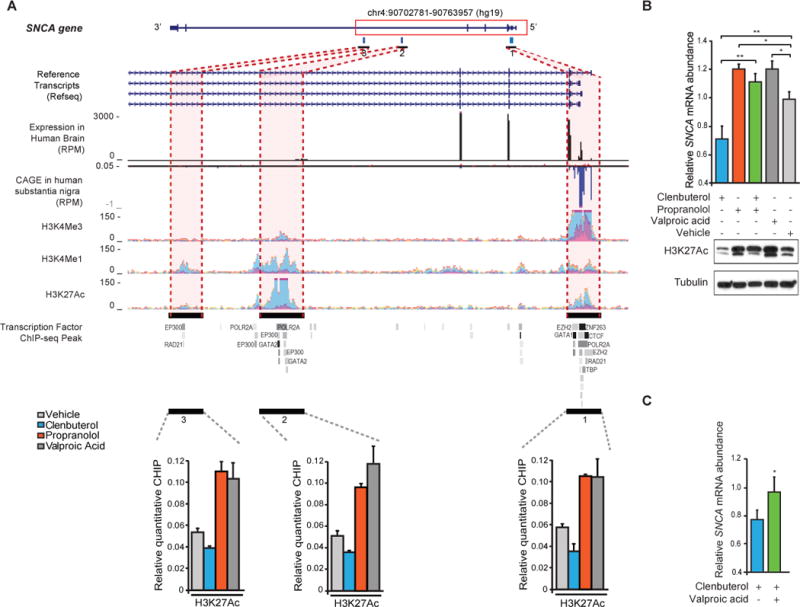
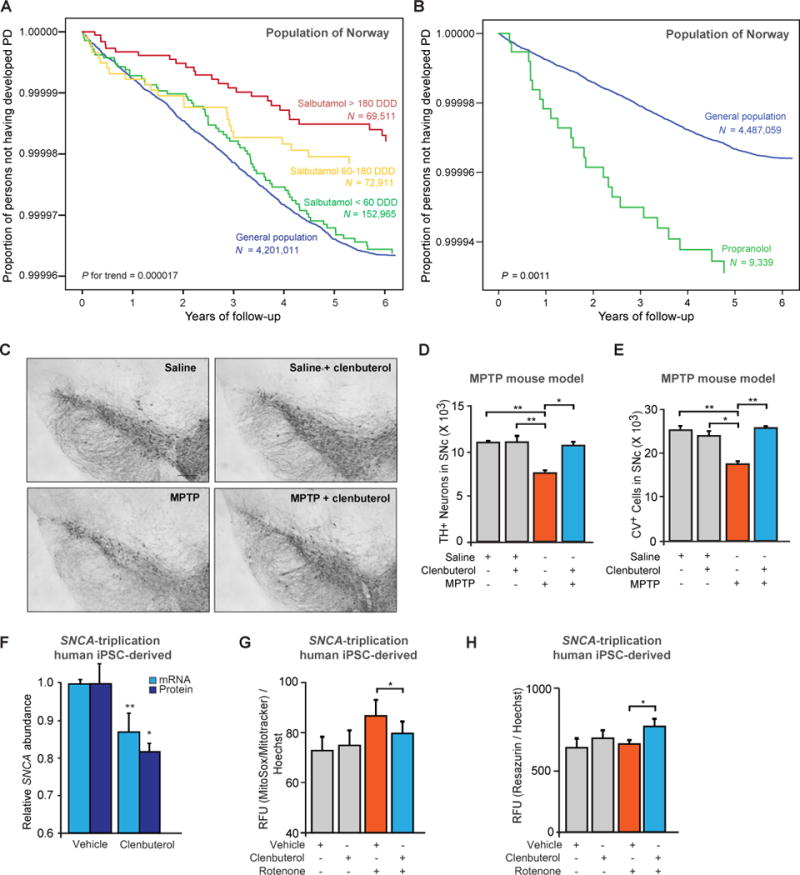
Copy number mutations implicate excess production of α-synuclein as a possibly causative factor in Parkinson's disease (PD). Using an unbiased screen targeting endogenous gene expression, we discovered that the β2-adrenoreceptor (β2AR) is a regulator of the α-synuclein gene (SNCA). β2AR ligands modulate SNCA transcription through histone 3 lysine 27 acetylation of its promoter and enhancers. Over 11 years of follow-up in 4 million Norwegians, the β2AR agonist salbutamol, a brain-penetrant asthma medication, was associated with reduced risk of developing PD (rate ratio, 0.66; 95% confidence interval, 0.58 to 0.76). Conversely, a β2AR antagonist correlated with increased risk. β2AR activation protected model mice and patient-derived cells. Thus, β2AR is linked to transcription of α-synuclein and risk of PD in a ligand-specific fashion and constitutes a potential target for therapies.
Copyright © 2017, American Association for the Advancement of Science.
Figures
Comment in
-
Finding a new purpose for old drugs.Science. 2017 Sep 1;357(6354):869-870. doi: 10.1126/science.aao2992. Science. 2017. PMID: 28860370 No abstract available.
-
Parkinson disease: Asthma drug could protect against PD.Nat Rev Neurol. 2017 Nov;13(11):640-641. doi: 10.1038/nrneurol.2017.134. Epub 2017 Sep 15. Nat Rev Neurol. 2017. PMID: 28914888 No abstract available.
-
Repurposing drugs to treat neurological diseases.J Neurol. 2018 Feb;265(2):446-448. doi: 10.1007/s00415-018-8732-z. J Neurol. 2018. PMID: 29322257 Free PMC article. No abstract available.
-
Identification of new α-synuclein regulator by nontraditional drug development pipeline.Mov Disord. 2018 Mar;33(3):402. doi: 10.1002/mds.27278. Epub 2018 Jan 11. Mov Disord. 2018. PMID: 29322595 Free PMC article. No abstract available.
References
-
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. - PubMed
-
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Annals of neurology. 2004;55:174–179. - PubMed
-
- Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. 2004;62:1835–1838. - PubMed
-
- Pihlstrom L, Toft M. Genetic variability in SNCA and Parkinson's disease. Neurogenetics. 2011;12:283–293. - PubMed
-
- Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, Mollenhauer B, Bresnick EH, Schlossmacher MG. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10907–10912. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
