

A novel rapid and reproducible flow cytometric method for optimization of transfection efficiency in cells
- PMID: 28863132
- PMCID: PMC5580984
- DOI: 10.1371/journal.pone.0182941
A novel rapid and reproducible flow cytometric method for optimization of transfection efficiency in cells
Abstract
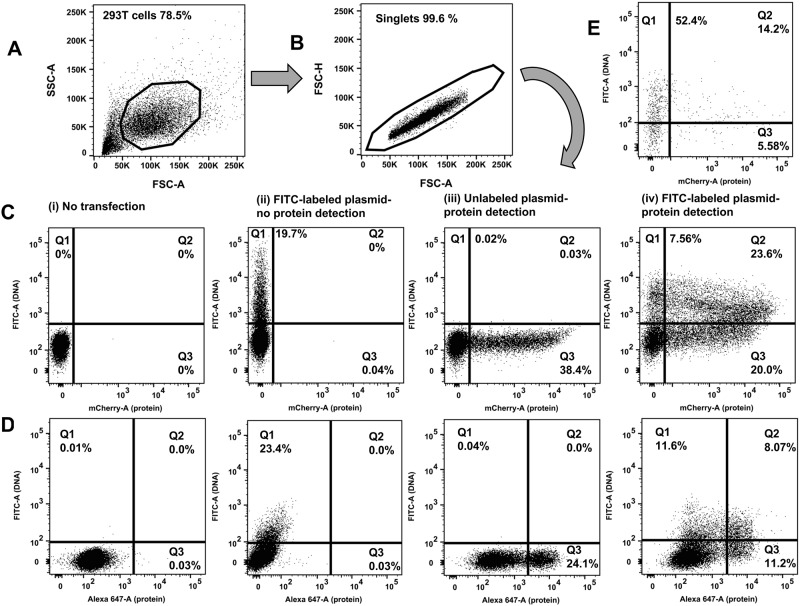
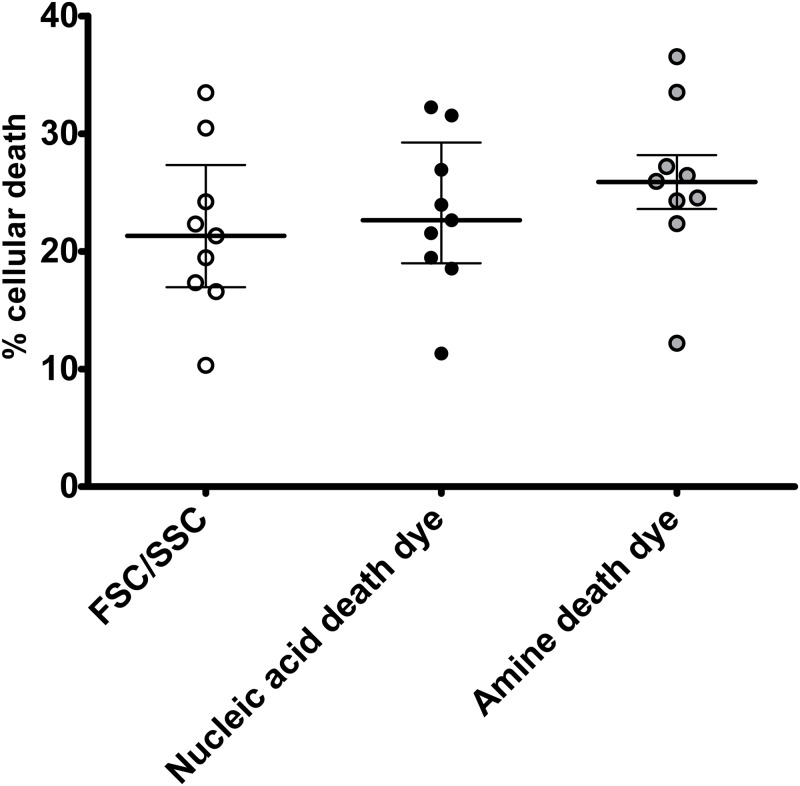
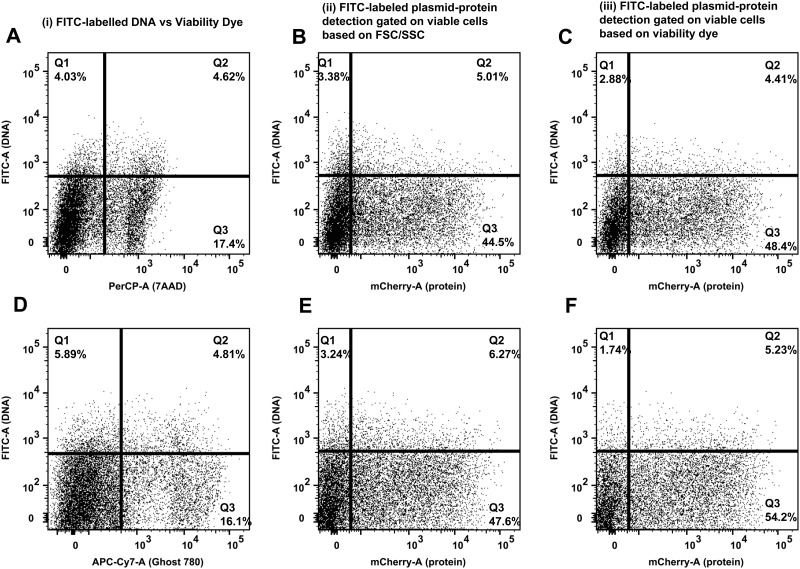
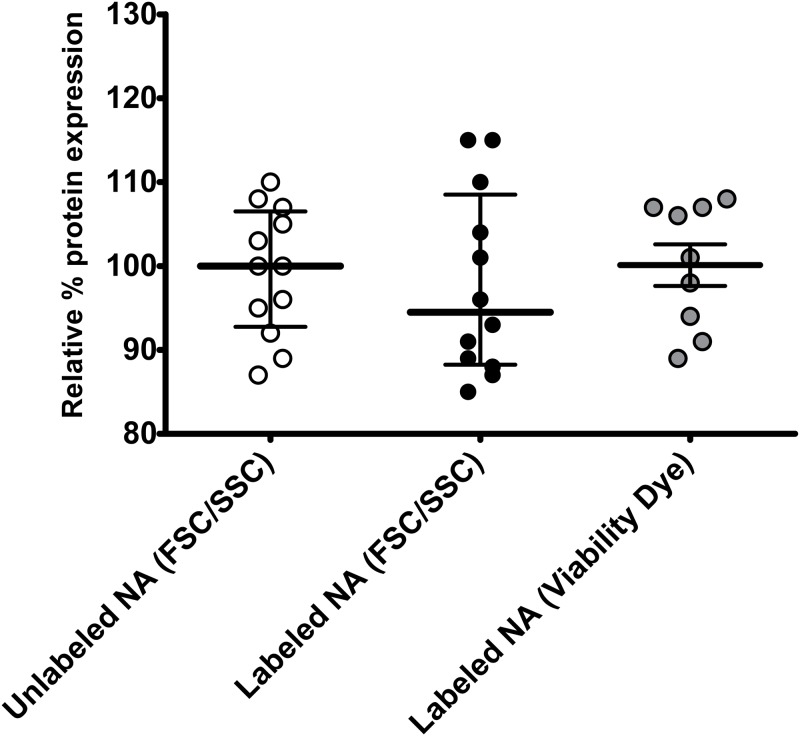
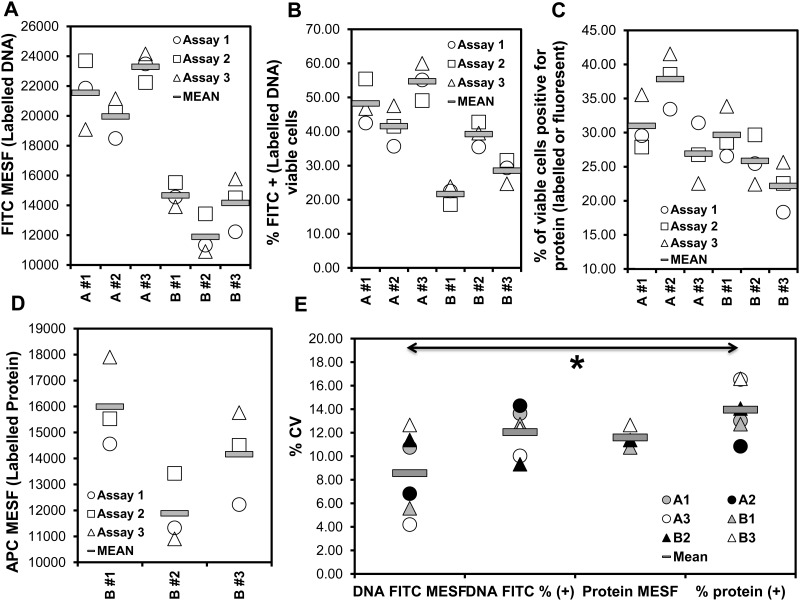
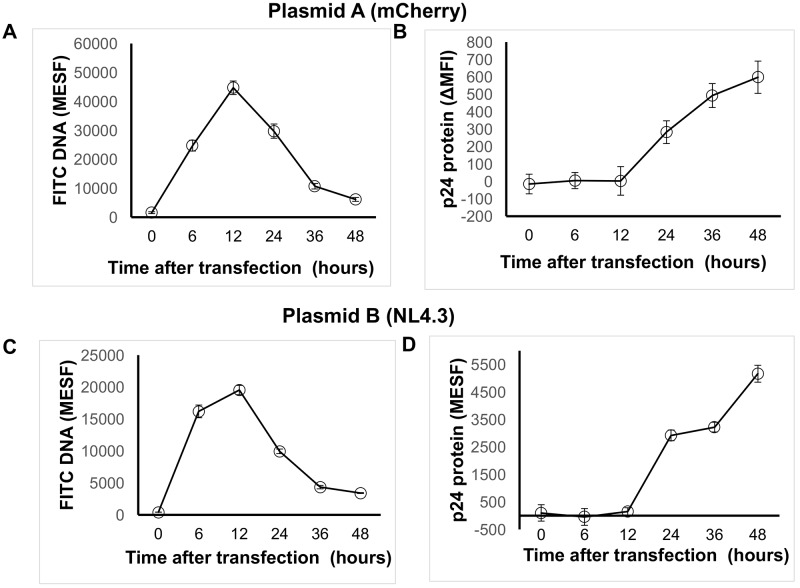
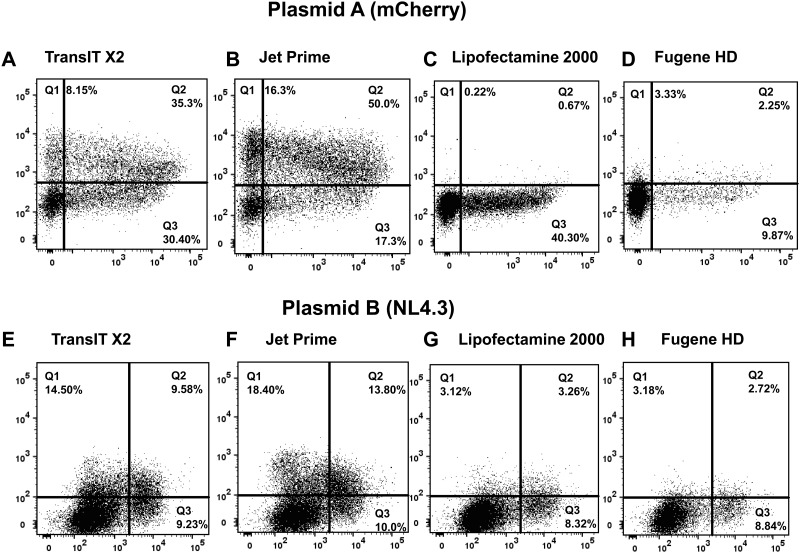
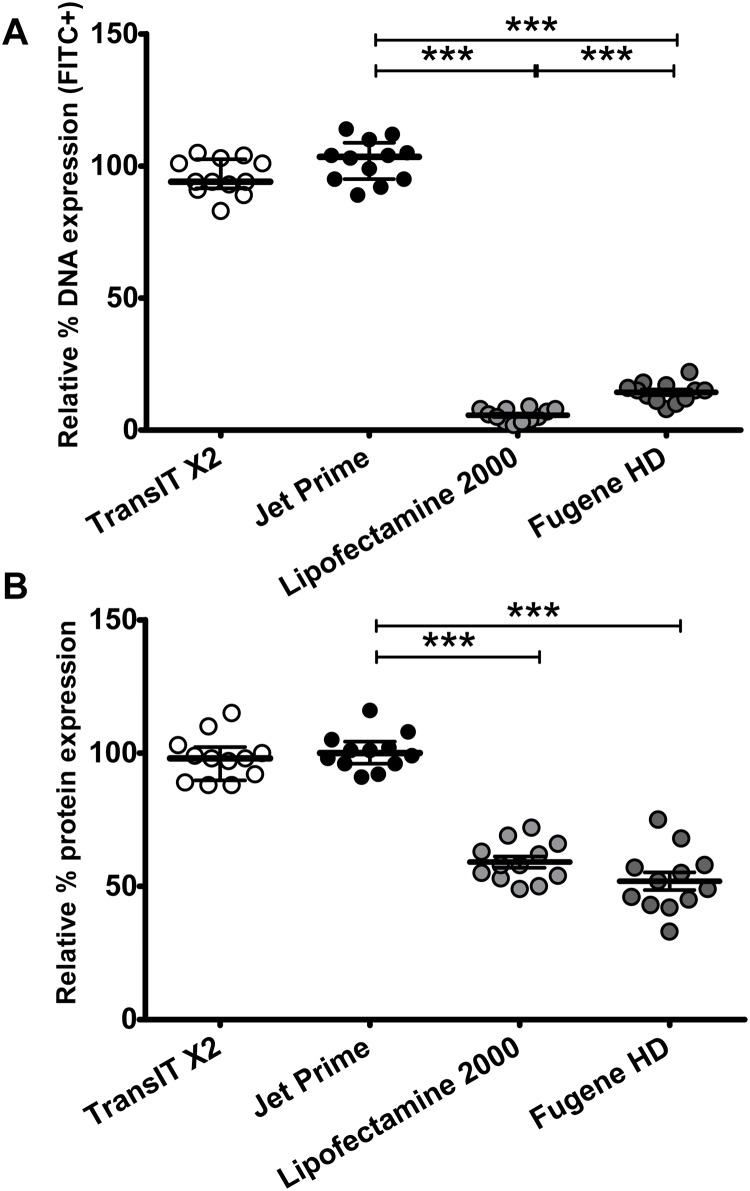
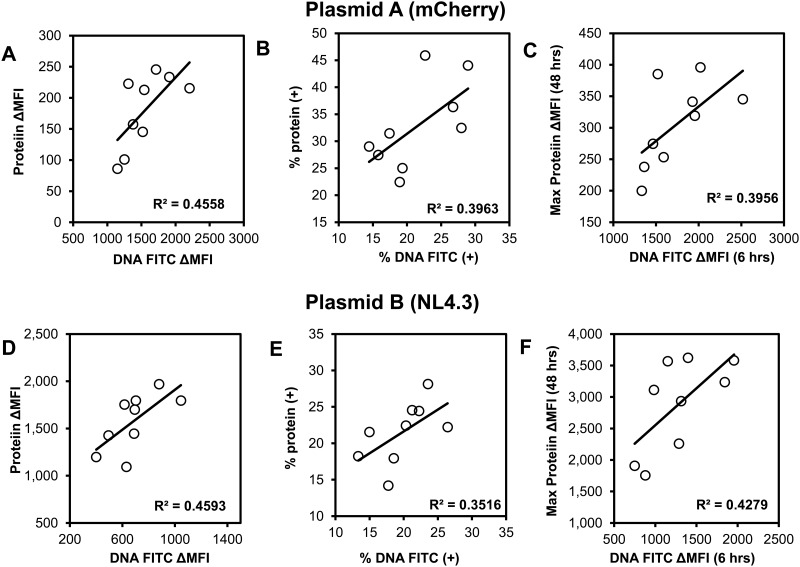
Transfection is one of the most frequently used techniques in molecular biology that is also applicable for gene therapy studies in humans. One of the biggest challenges to investigate the protein function and interaction in gene therapy studies is to have reliable monospecific detection reagents, particularly antibodies, for all human gene products. Thus, a reliable method that can optimize transfection efficiency based on not only expression of the target protein of interest but also the uptake of the nucleic acid plasmid, can be an important tool in molecular biology. Here, we present a simple, rapid and robust flow cytometric method that can be used as a tool to optimize transfection efficiency at the single cell level while overcoming limitations of prior established methods that quantify transfection efficiency. By using optimized ratios of transfection reagent and a nucleic acid (DNA or RNA) vector directly labeled with a fluorochrome, this method can be used as a tool to simultaneously quantify cellular toxicity of different transfection reagents, the amount of nucleic acid plasmid that cells have taken up during transfection as well as the amount of the encoded expressed protein. Finally, we demonstrate that this method is reproducible, can be standardized and can reliably and rapidly quantify transfection efficiency, reducing assay costs and increasing throughput while increasing data robustness.
Conflict of interest statement
Figures
References
-
- Stoll SM, Calos MP. Extrachromosomal plasmid vectors for gene therapy. Curr Opin Mol Ther. 2002;4(4):299–305. . - PubMed
-
- Kim TK, Eberwine JH. Mammalian cell transfection: the present and the future. Anal Bioanal Chem. 2010;397(8):3173–8. doi: 10.1007/s00216-010-3821-6 . - DOI - PMC - PubMed
-
- Zhou ZL, Sun XX, Ma J, Man CH, Wong AS, Leung AY, et al. Mechanical oscillations enhance gene delivery into suspended cells. Sci Rep. 2016;6:22824 doi: 10.1038/srep22824 . - DOI - PMC - PubMed
-
- Wang L, Zhang J, Zhang R, Xue F, Sun Y, Han X. Limitation in use of luciferase reporter genes for 3'-untranslated region analysis. Biotechnol Lett. 2007;29(11):1691–6. doi: 10.1007/s10529-007-9448-9 . - DOI - PubMed
-
- Wilson SM, Bacic A. Preparation of plant cells for transmission electron microscopy to optimize immunogold labeling of carbohydrate and protein epitopes. Nat Protoc. 2012;7(9):1716–27. doi: 10.1038/nprot.2012.096 . - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
