

Recurrent Tumor Cell-Intrinsic and -Extrinsic Alterations during MAPKi-Induced Melanoma Regression and Early Adaptation
- PMID: 28864476
- PMCID: PMC6668729
- DOI: 10.1158/2159-8290.CD-17-0401
Recurrent Tumor Cell-Intrinsic and -Extrinsic Alterations during MAPKi-Induced Melanoma Regression and Early Adaptation
Abstract
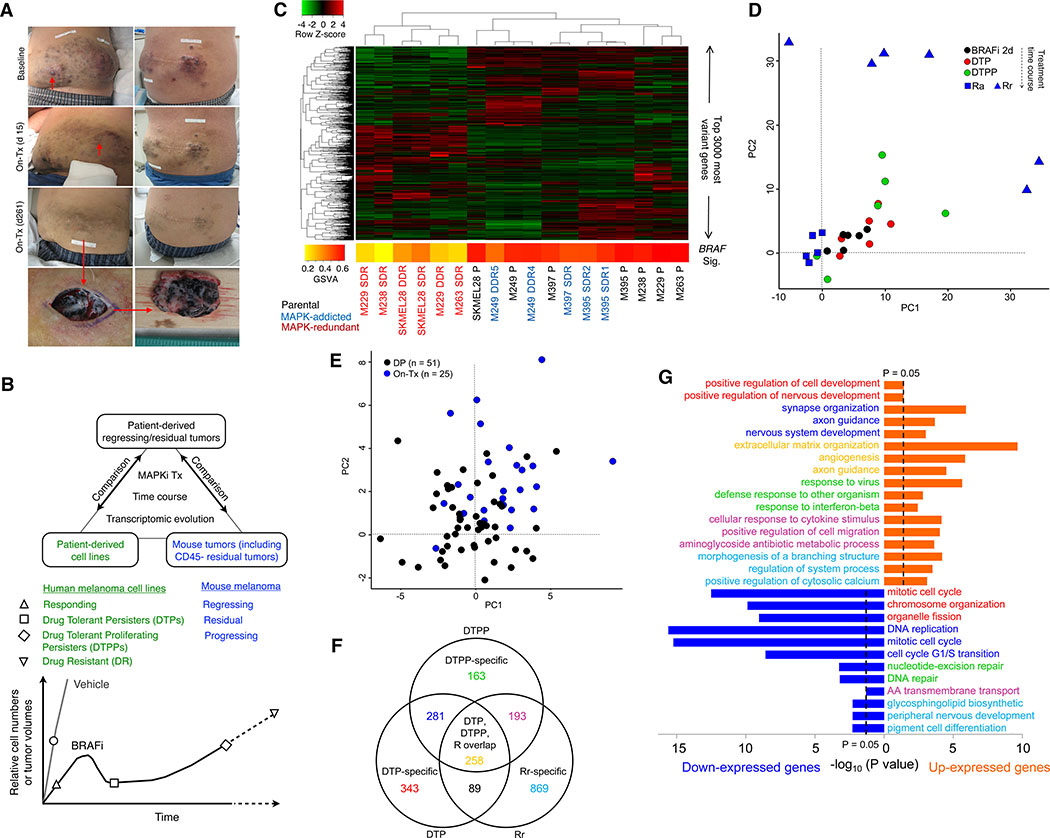
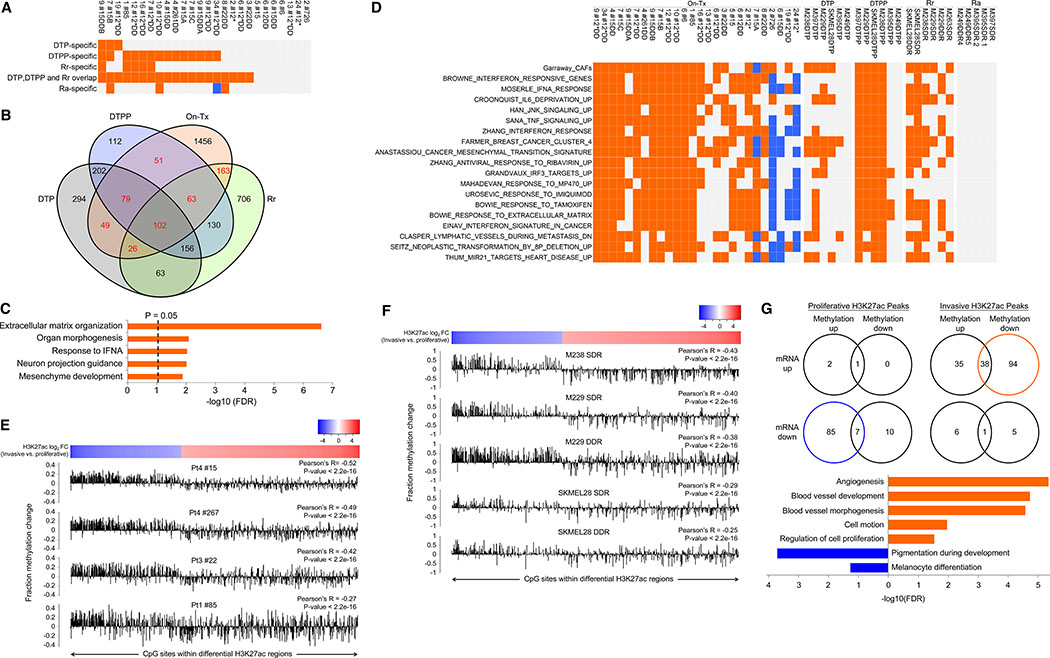
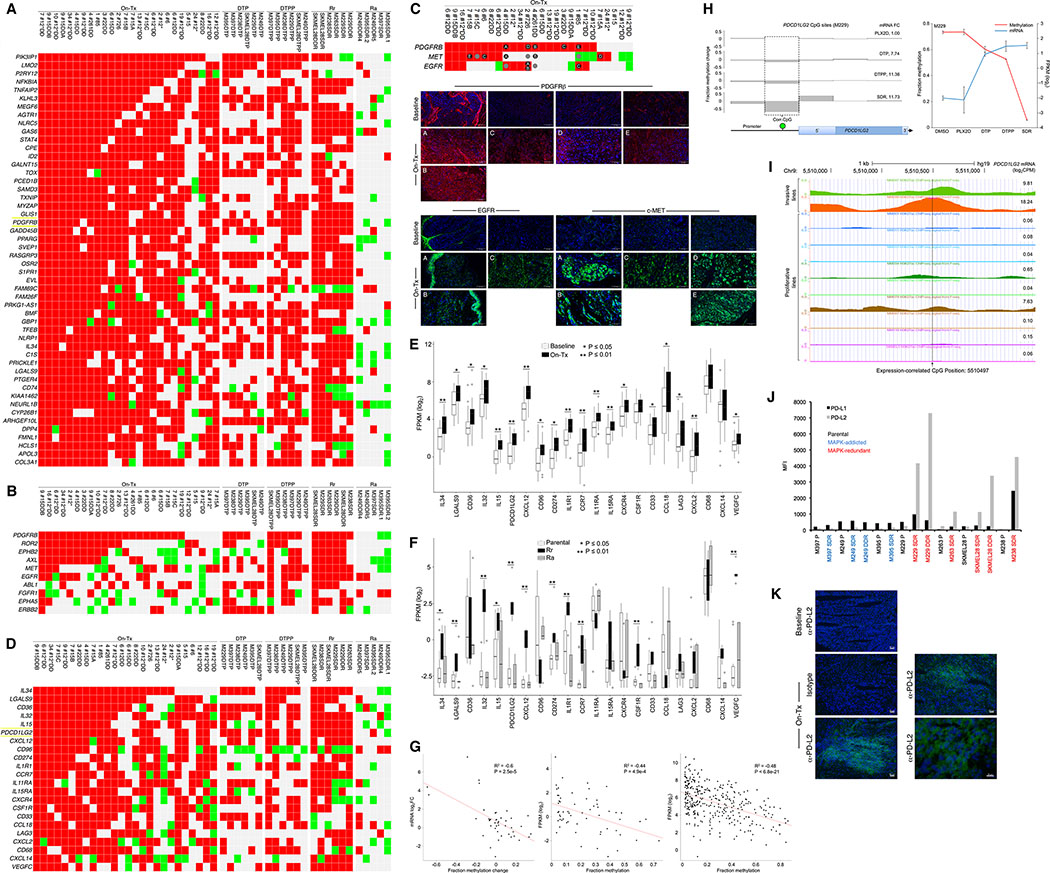
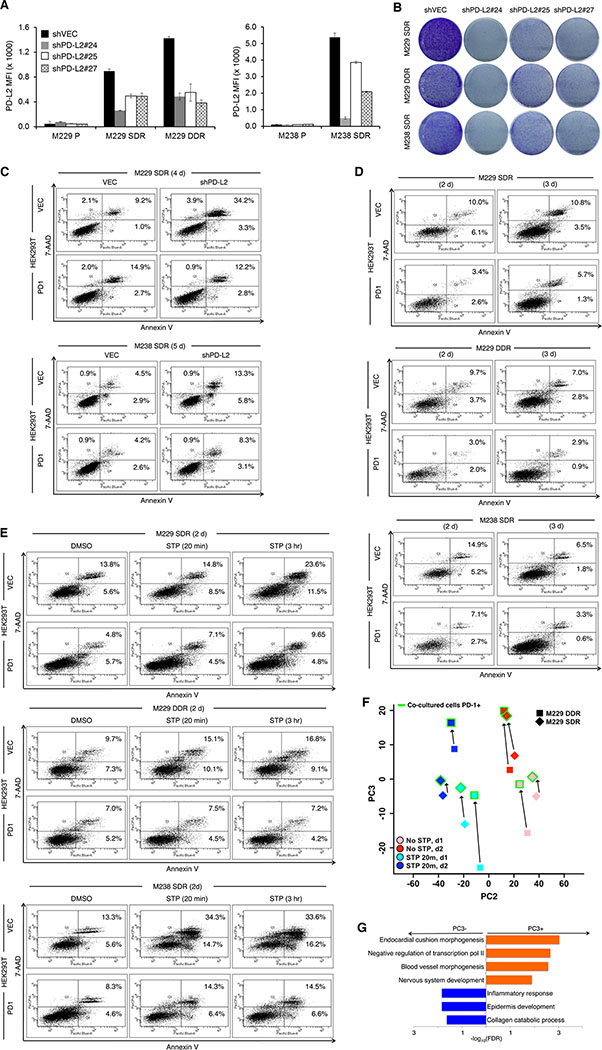
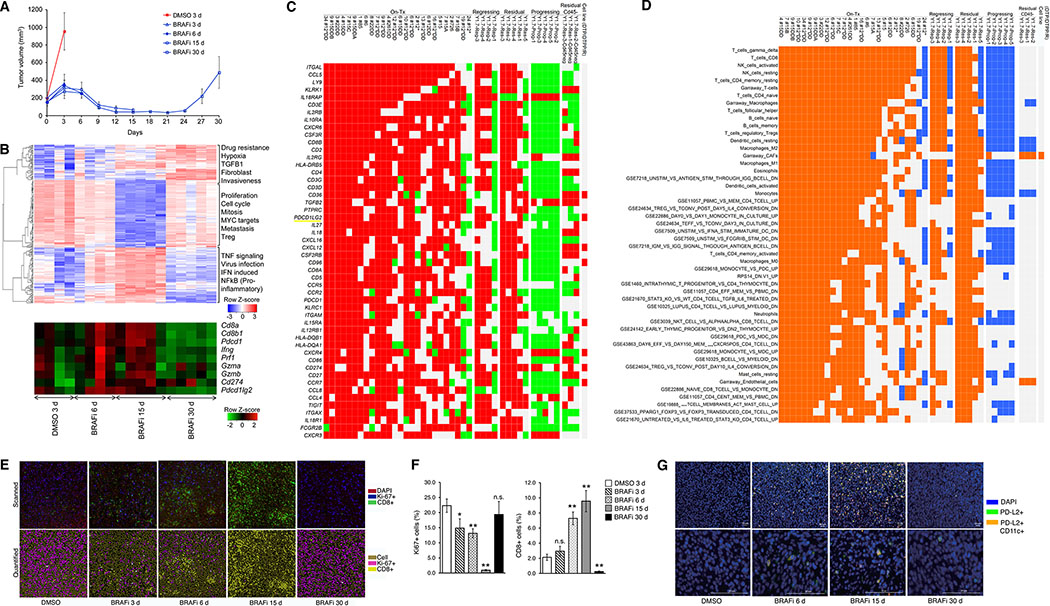
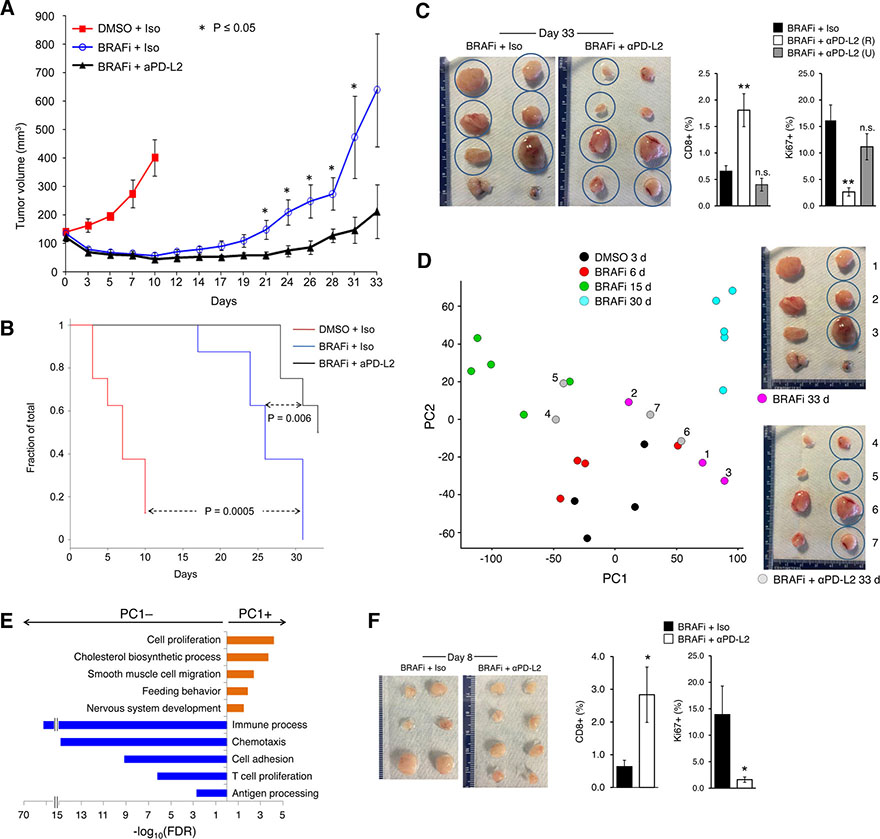
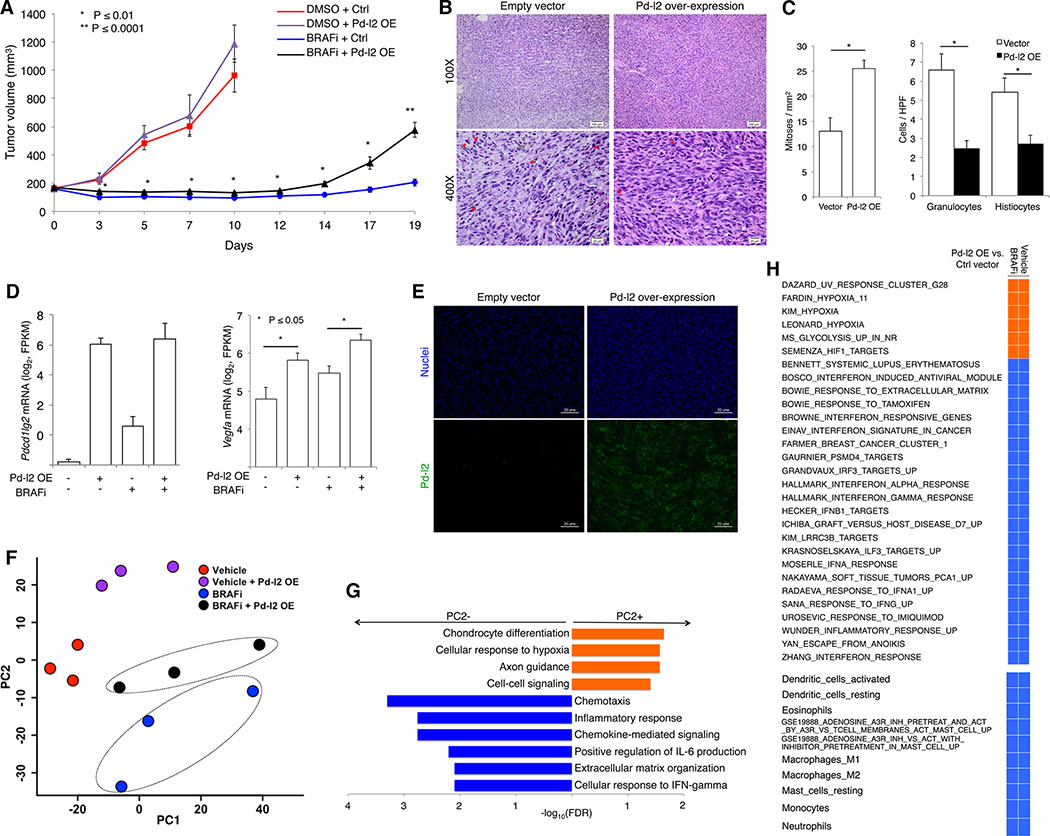
Treatment of advanced BRAFV600-mutant melanoma using a BRAF inhibitor or its combination with a MEK inhibitor typically elicits partial responses. We compared the transcriptomes of patient-derived tumors regressing on MAPK inhibitor (MAPKi) therapy against MAPKi-induced temporal transcriptomic states in human melanoma cell lines or murine melanoma in immune-competent mice. Despite heterogeneous dynamics of clinical tumor regression, residual tumors displayed highly recurrent transcriptomic alterations and enriched processes, which were also observed in MAPKi-selected cell lines (implying tumor cell-intrinsic reprogramming) or in bulk mouse tumors (and the CD45-negative or CD45-positive fractions, implying tumor cell-intrinsic or stromal/immune alterations, respectively). Tumor cell-intrinsic reprogramming attenuated MAPK dependency, while enhancing mesenchymal, angiogenic, and IFN-inflammatory features and growth/survival dependence on multi-RTKs and PD-L2. In the immune compartment, PD-L2 upregulation in CD11c+ immunocytes drove the loss of T-cell inflammation and promoted BRAFi resistance. Thus, residual melanoma early on MAPKi therapy already displays potentially exploitable adaptive transcriptomic, epigenomic, immune-regulomic alterations.Significance: Incomplete MAPKi-induced melanoma regression results in transcriptome/methylome-wide reprogramming and MAPK-redundant escape. Although regressing/residual melanoma is highly T cell-inflamed, stromal adaptations, many of which are tumor cell-driven, could suppress/eliminate intratumoral T cells, reversing tumor regression. This catalog of recurrent alterations helps identify adaptations such as PD-L2 operative tumor cell intrinsically and/or extrinsically early on therapy. Cancer Discov; 7(11); 1248-65. ©2017 AACR.See related commentary by Haq, p. 1216This article is highlighted in the In This Issue feature, p. 1201.
©2017 American Association for Cancer Research.
Conflict of interest statement
Authors declare no conflicts of interest.
Figures
Comment in
-
Trapping Cancers as They Adapt to Survive.Cancer Discov. 2017 Nov;7(11):1216-1217. doi: 10.1158/2159-8290.CD-17-0976. Cancer Discov. 2017. PMID: 29097618
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
