

Defects at the crossroads of GABAergic signaling in generalized genetic epilepsies
- PMID: 28865303
- PMCID: PMC6112605
- DOI: 10.1016/j.eplepsyres.2017.08.013
Defects at the crossroads of GABAergic signaling in generalized genetic epilepsies
Abstract
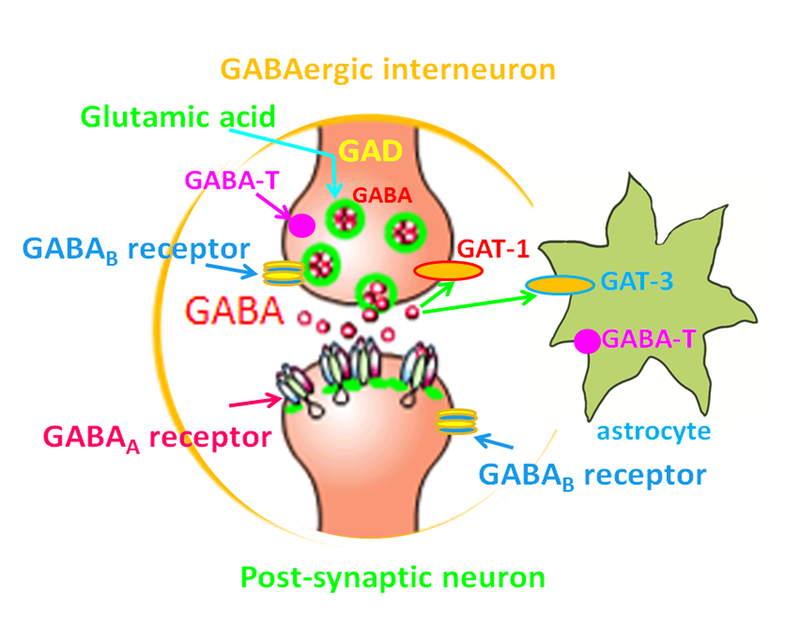
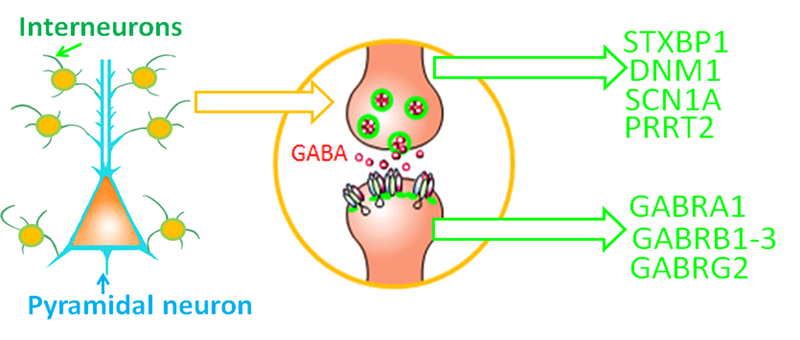
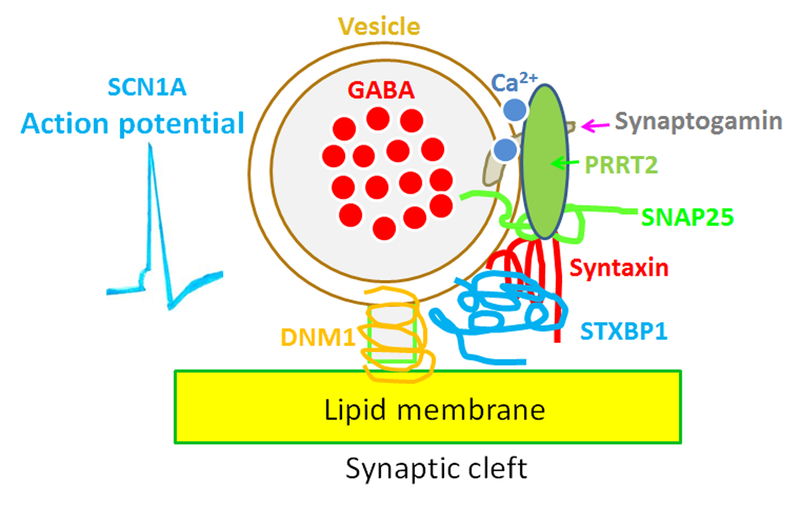
Seizure disorders are very common and affect 3% of the general population. The recurrent unprovoked seizures that are also called epilepsies are highly diverse as to both underlying genetic basis and clinic presentations. Recent genetic advances and sequencing technologies indicate that many epilepsies previously thought to be without known causes, or idiopathic generalized epilepsies (IGEs), are virtually genetic epilepsy as they are caused by genetic variations. IGEs are estimated to account for ∼15-20% of all epilepsies. Initially IGEs were primarily considered channelopathies, because the first genetic defects identified in IGEs involved ion channel genes. However, new findings indicate that mutations in many non ion channel genes are also involved in addition to those in ion channel genes. Interestingly, mutations in many genes associated with epilepsy affect GABAergic signaling, a major biological pathway in epilepsy. Additionally, many antiepileptic drugs work via enhancing GABAergic signaling. Hence, the review will focus on the mutations that impair GABAergic signaling and selectively discuss the newly identified STXBP1, PRRT2, and DNM1 in addition to those long-established epilepsy ion channel genes that also impair GABAergic signaling like SCN1A and GABAA receptor subunit genes. GABAergic signaling includes the pre- and post- synaptic mechanisms. Some mutations, such as STXBP1, PRRT2, DNM1, and SCN1A, impair GABAergic signaling mainly via pre-synaptic mechanisms while those mutations in GABAA receptor subunit genes impair GABAergic signaling via post-synaptic mechanisms. Nevertheless, these findings suggest impaired GABAergic signaling is a converging pathway of defects for many ion channel or non ion channel mutations associated with genetic epilepsies.
Keywords: Epilepsy; GABAergic signaling; Ion channels; Mutations; Non ion channels; Vesicles.
Copyright © 2017 Elsevier B.V. All rights reserved.
Figures
References
-
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli NS, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin TL, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR, 2013. De novo mutations in epileptic encephalopathies. Nature 501, 217–221. - PMC - PubMed
-
- Anderson E, Berkovic S, Dulac O, Gardiner M, Jain S, Laue FM, Lindhout D, Noebels J, Ottman R, Scaramelli A, Serratosa J, Steinlein O, Avanzini G, Bailey-Wilson J, Cardon L, Fischbach R, Gwinn-Hardy K, Leppert M, Ott J, Lindblad-Toh K, Weiss K, 2002. ILAE genetics commission conference report: molecular analysis of complex genetic epilepsies. Epilepsia 43, 1262–1267. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
