

Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder
- PMID: 28867142
- PMCID: PMC5590950
- DOI: 10.1016/j.ajhg.2017.07.016
Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder
Abstract
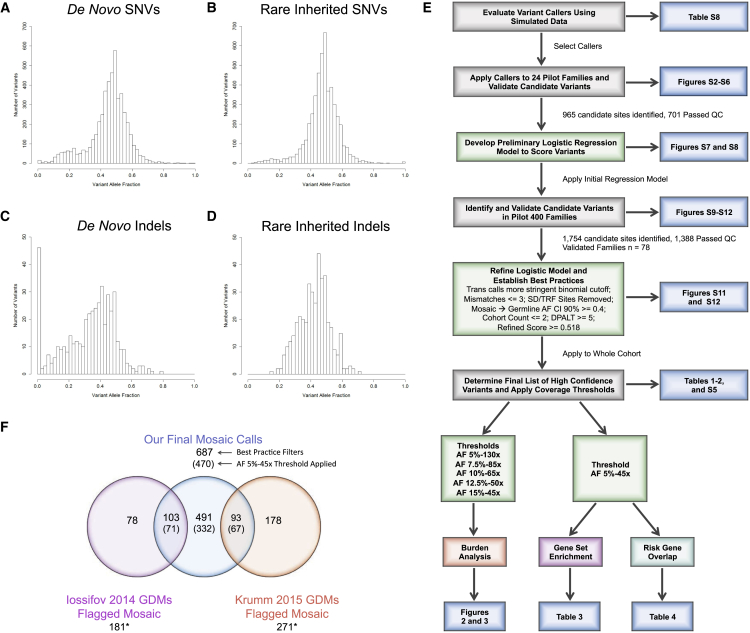
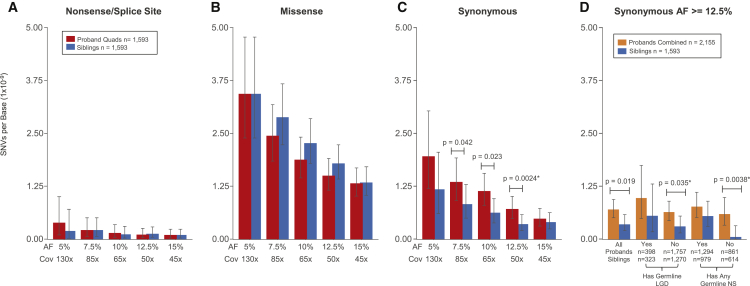
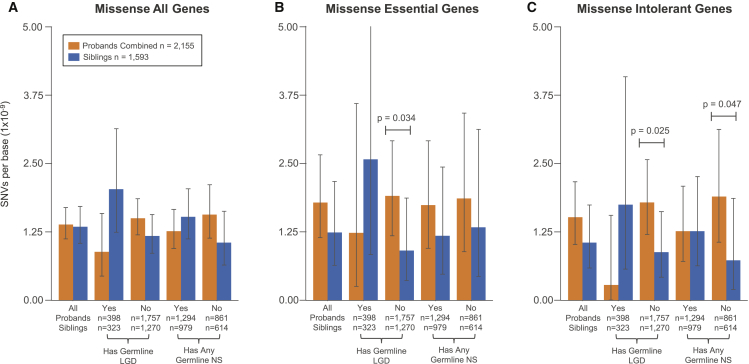
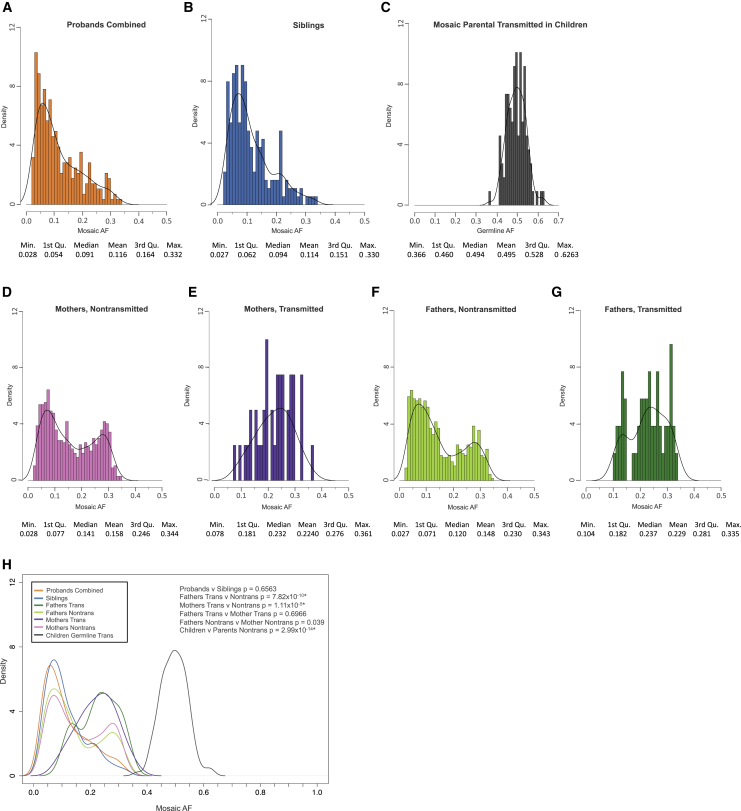
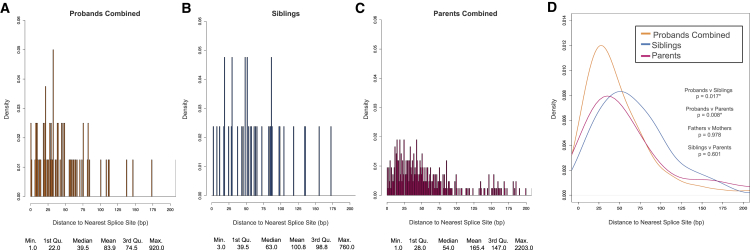
Genetic risk factors for autism spectrum disorder (ASD) have yet to be fully elucidated. Postzygotic mosaic mutations (PMMs) have been implicated in several neurodevelopmental disorders and overgrowth syndromes. By leveraging whole-exome sequencing data on a large family-based ASD cohort, the Simons Simplex Collection, we systematically evaluated the potential role of PMMs in autism risk. Initial re-evaluation of published single-nucleotide variant (SNV) de novo mutations showed evidence consistent with putative PMMs for 11% of mutations. We developed a robust and sensitive SNV PMM calling approach integrating complementary callers, logistic regression modeling, and additional heuristics. In our high-confidence call set, we identified 470 PMMs in children, increasing the proportion of mosaic SNVs to 22%. Probands have a significant burden of synonymous PMMs and these mutations are enriched for computationally predicted impacts on splicing. Evidence of increased missense PMM burden was not seen in the full cohort. However, missense burden signal increased in subcohorts of families where probands lacked nonsynonymous germline mutations, especially in genes intolerant to mutations. Parental mosaic mutations that were transmitted account for 6.8% of the presumed de novo mutations in the children. PMMs were identified in previously implicated high-confidence neurodevelopmental disorder risk genes, such as CHD2, CTNNB1, SCN2A, and SYNGAP1, as well as candidate risk genes with predicted functions in chromatin remodeling or neurodevelopment, including ACTL6B, BAZ2B, COL5A3, SSRP1, and UNC79. We estimate that PMMs potentially contribute risk to 3%-4% of simplex ASD case subjects and future studies of PMMs in ASD and related disorders are warranted.
Keywords: autism spectrum disorder; exome; mosaicism; mutation; neurodevelopment; postzygotic; somatic; splicing.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
