

Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses
- PMID: 28867387
- PMCID: PMC5604429
- DOI: 10.1016/j.chom.2017.07.019
Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses
Abstract
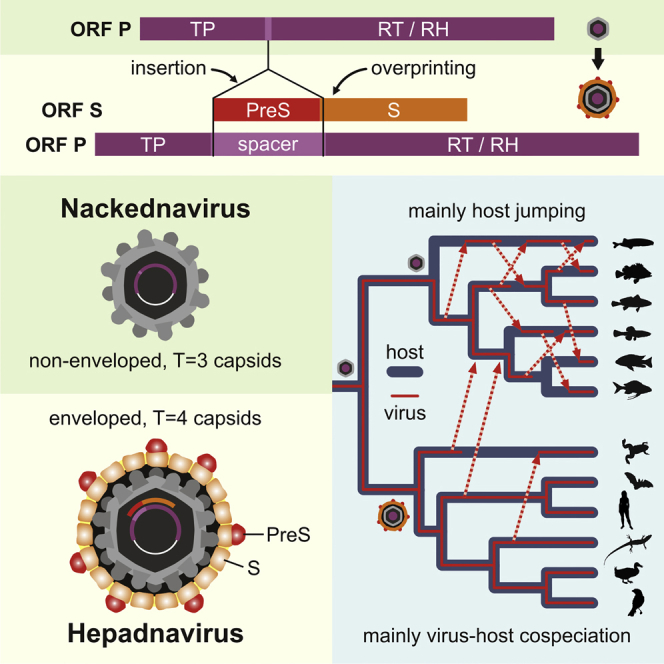
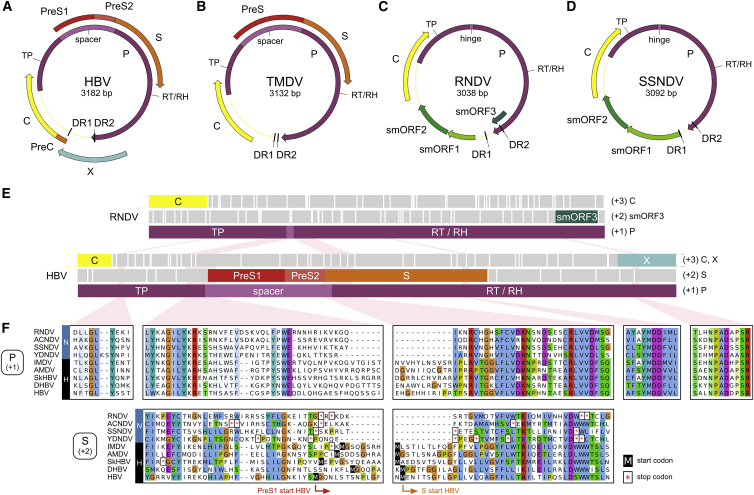
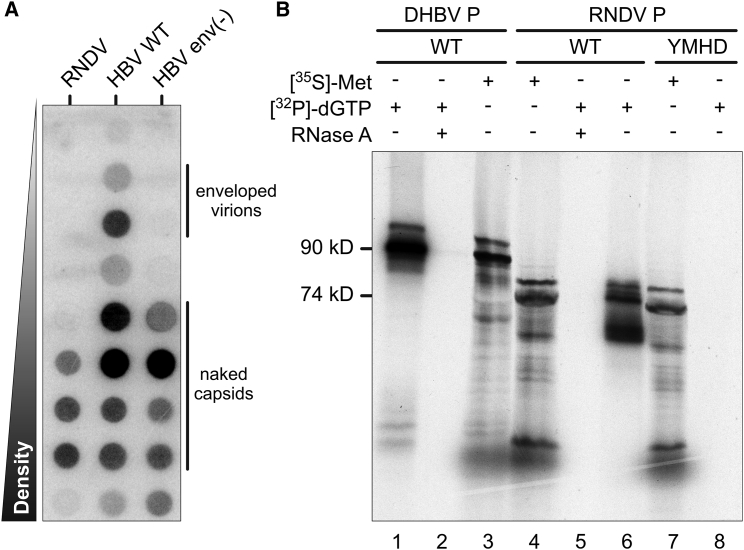
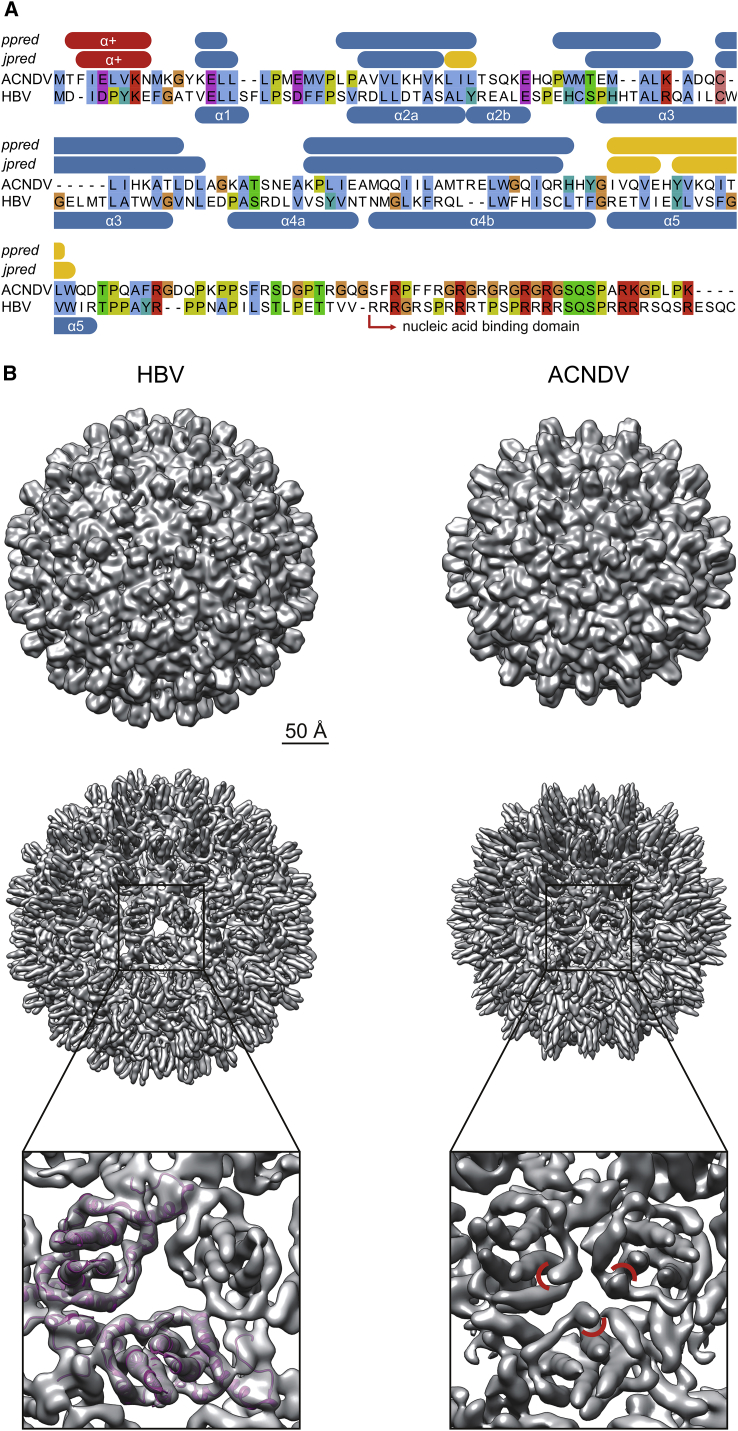
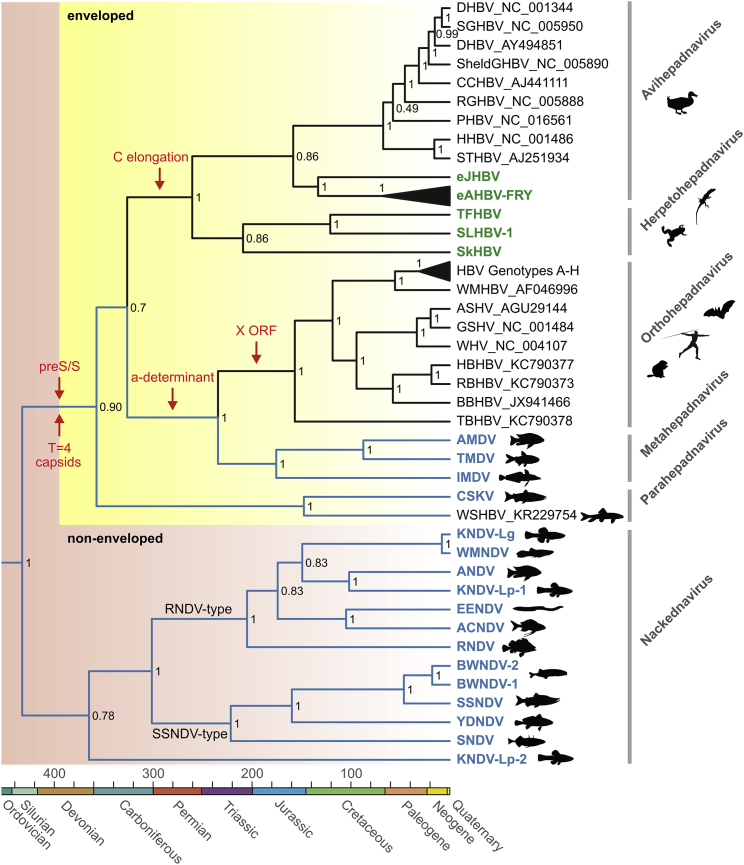
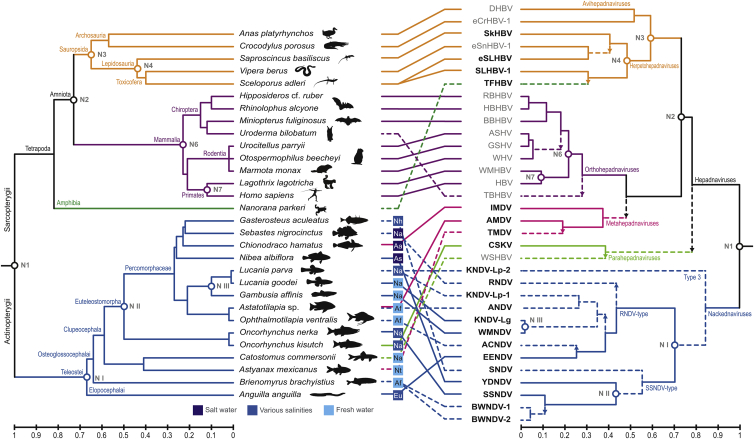
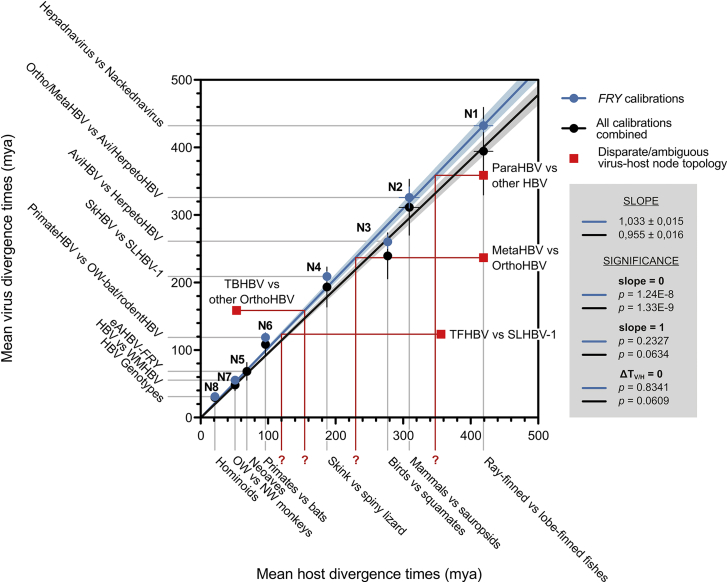
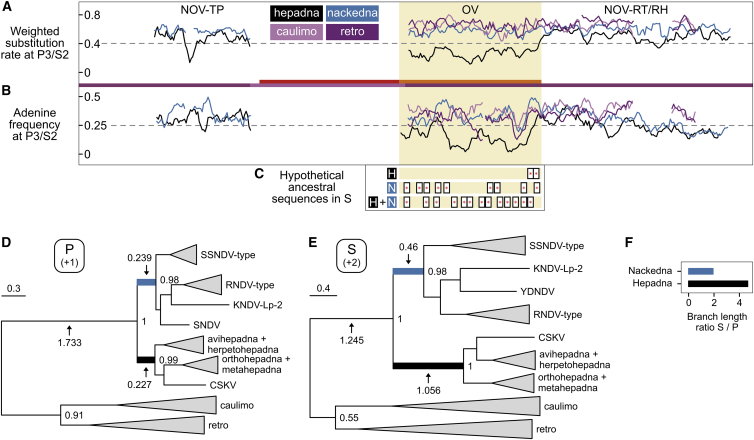
Hepatitis B viruses (HBVs), which are enveloped viruses with reverse-transcribed DNA genomes, constitute the family Hepadnaviridae. An outstanding feature of HBVs is their streamlined genome organization with extensive gene overlap. Remarkably, the ∼1,100 bp open reading frame (ORF) encoding the envelope proteins is fully nested within the ORF of the viral replicase P. Here, we report the discovery of a diversified family of fish viruses, designated nackednaviruses, which lack the envelope protein gene, but otherwise exhibit key characteristics of HBVs including genome replication via protein-primed reverse-transcription and utilization of structurally related capsids. Phylogenetic reconstruction indicates that these two virus families separated more than 400 million years ago before the rise of tetrapods. We show that HBVs are of ancient origin, descending from non-enveloped progenitors in fishes. Their envelope protein gene emerged de novo, leading to a major transition in viral lifestyle, followed by co-evolution with their hosts over geologic eras.
Keywords: hepadnaviruses; hepatitis B virus; overlapping open reading frames; viral gene evolution; virus discovery; virus origins; virus-host long-term co-evolution.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Abascal F., Zardoya R., Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. - PubMed
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Andrews, S. (2010). FastQC. A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
