

Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation
- PMID: 28868155
- PMCID: PMC5576364
- DOI: 10.1038/npjgenmed.2016.36
Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation
Abstract
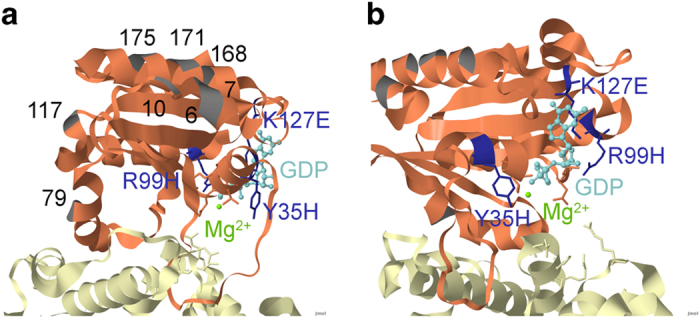
Genomic sequence interpretation can miss clinically relevant missense variants for several reasons. Rare missense variants are numerous in the exome and difficult to prioritise. Affected genes may also not have existing disease association. To improve variant prioritisation, we leverage population exome data to identify intragenic missense-depleted regions (MDRs) genome-wide that may be important in disease. We then use missense depletion analyses to help prioritise undiagnosed disease exome variants. We demonstrate application of this strategy to identify a novel gene association for human brain malformation. We identified de novo missense variants that affect the GDP/GTP-binding site of ARF1 in three unrelated patients. Corresponding functional analysis suggests ARF1 GDP/GTP-activation is affected by the specific missense mutations associated with heterotopia. These findings expand the genetic pathway underpinning neurologic disease that classically includes FLNA. ARF1 along with ARFGEF2 add further evidence implicating ARF/GEFs in the brain. Using functional ontology, top MDR-containing genes were highly enriched for nucleotide-binding function, suggesting these may be candidates for human disease. Routine consideration of MDR in the interpretation of exome data for rare diseases may help identify strong genetic factors for many severe conditions, infertility/reduction in reproductive capability, and embryonic conditions contributing to preterm loss.
Keywords: ARF1; Exome sequencing; GDP/GTP; MDR; RAS superfamily; brain malformation; missense-depletion; nucleotide-binding; variant prioritization.
Conflict of interest statement
COMPETING INTERESTS The authors declare no conflict of interest.
Figures




References
Grants and funding
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- T32 GM008568/GM/NIGMS NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- K08 HL092970/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
