

Chromosome contacts in activated T cells identify autoimmune disease candidate genes
- PMID: 28870212
- PMCID: PMC5584004
- DOI: 10.1186/s13059-017-1285-0
Chromosome contacts in activated T cells identify autoimmune disease candidate genes
Abstract
Background: Autoimmune disease-associated variants are preferentially found in regulatory regions in immune cells, particularly CD4+ T cells. Linking such regulatory regions to gene promoters in disease-relevant cell contexts facilitates identification of candidate disease genes.
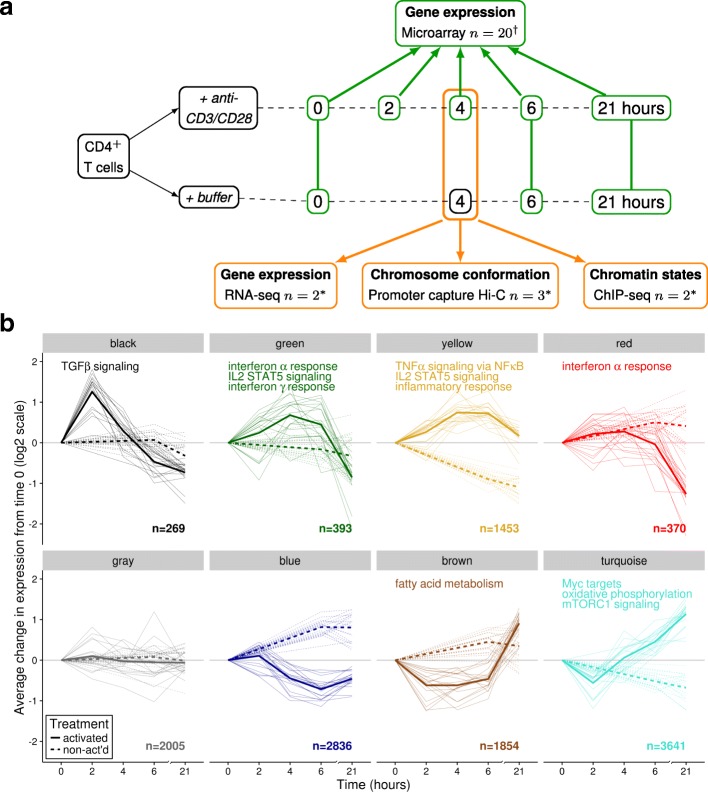
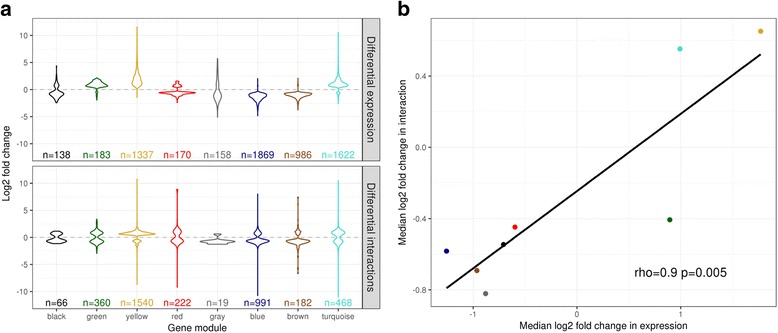
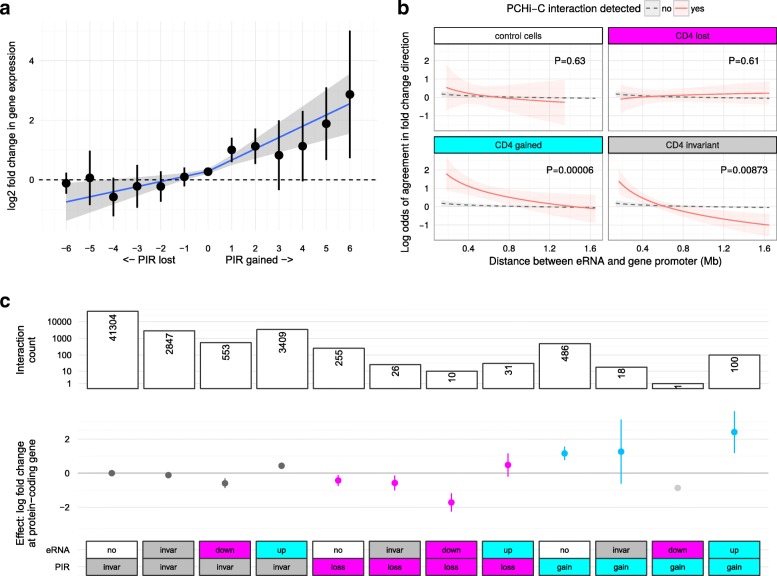
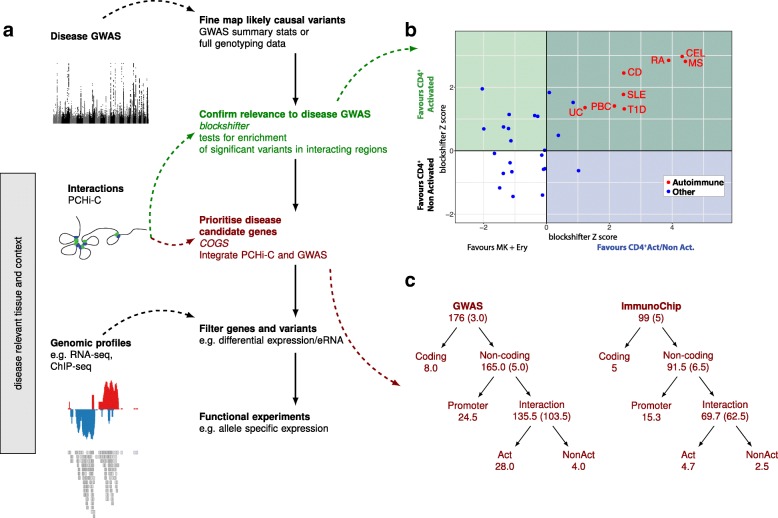
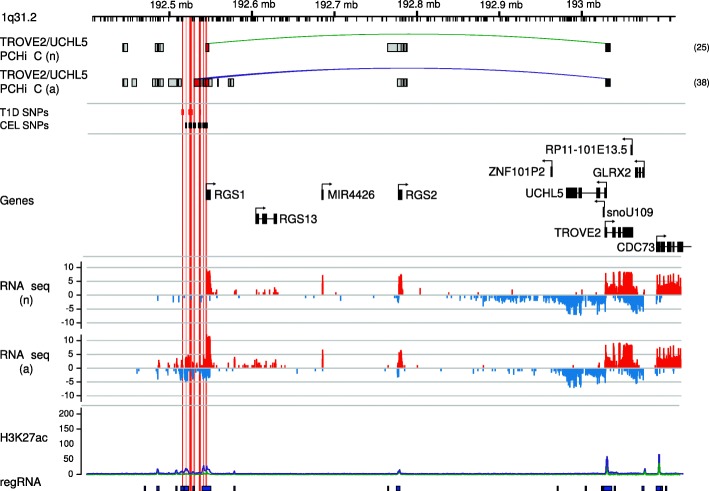
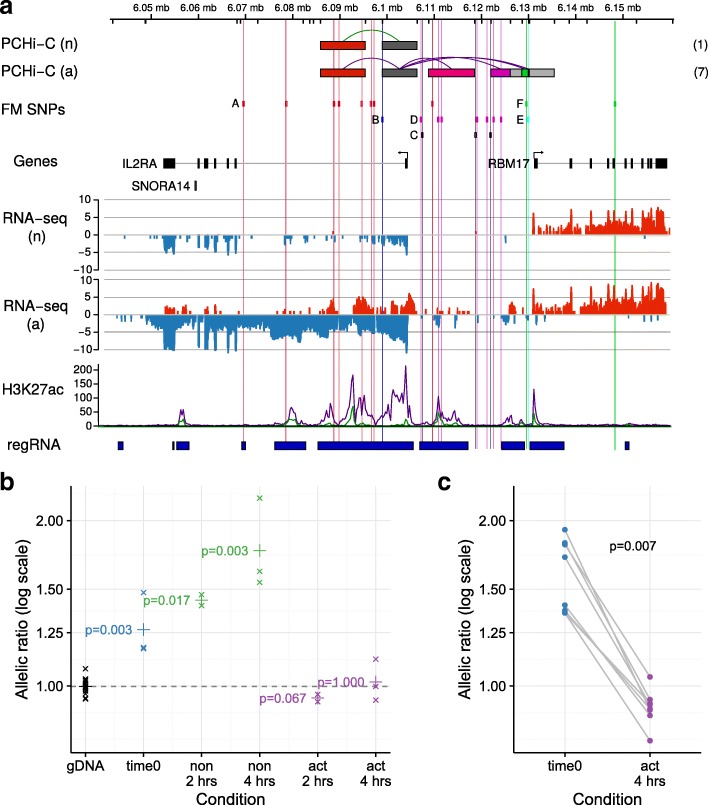
Results: Within 4 h, activation of CD4+ T cells invokes changes in histone modifications and enhancer RNA transcription that correspond to altered expression of the interacting genes identified by promoter capture Hi-C. By integrating promoter capture Hi-C data with genetic associations for five autoimmune diseases, we prioritised 245 candidate genes with a median distance from peak signal to prioritised gene of 153 kb. Just under half (108/245) prioritised genes related to activation-sensitive interactions. This included IL2RA, where allele-specific expression analyses were consistent with its interaction-mediated regulation, illustrating the utility of the approach.
Conclusions: Our systematic experimental framework offers an alternative approach to candidate causal gene identification for variants with cell state-specific functional effects, with achievable sample sizes.
Keywords: Autoimmune disease; CD4+ T cell activation; CD4+ T cells; Chromatin conformation; Genetics; Genome-wide association studies; Genomics.
Conflict of interest statement
Ethics approval and consent to participate
All samples and information were collected with written and signed informed consent. The study was approved by the local Peterborough and Fenland research ethics committee for the project entitled: ‘An investigation into genes and mechanisms based on genotype-phenotype correlations in type 1 diabetes and related diseases using peripheral blood mononuclear cells from volunteers that are part of the Cambridge BioResource project’ (05/Q0106/20). Experimental methods comply with the Helsinki Declaration.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
