

Neuron-derived CCL2 contributes to microglia activation and neurological decline in hepatic encephalopathy
- PMID: 28870240
- PMCID: PMC5584513
- DOI: 10.1186/s40659-017-0130-y
Neuron-derived CCL2 contributes to microglia activation and neurological decline in hepatic encephalopathy
Abstract
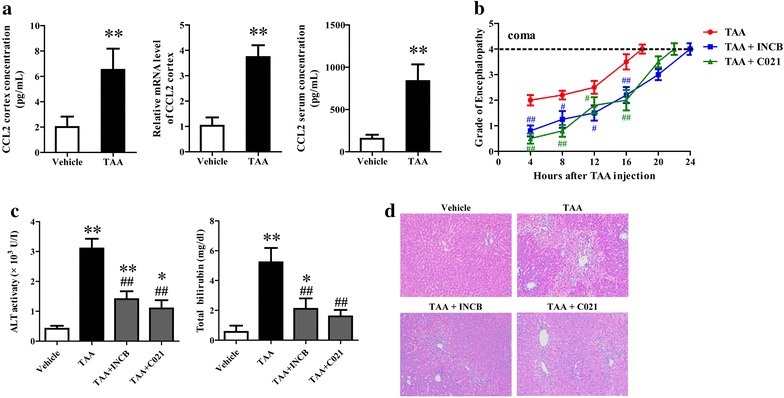
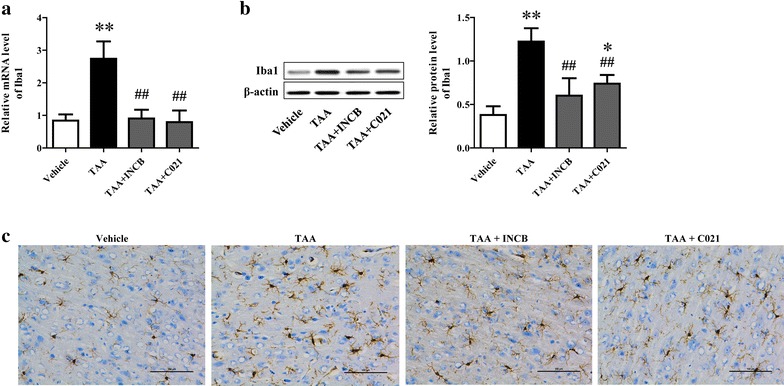
Background: CCL2 was up-regulated in neurons and involved in microglia activation and neurological decline in mice suffering from hepatic encephalopathy (HE). However, no data exist concerning the effect of neuron-derived CCL2 on microglia activation in vitro.
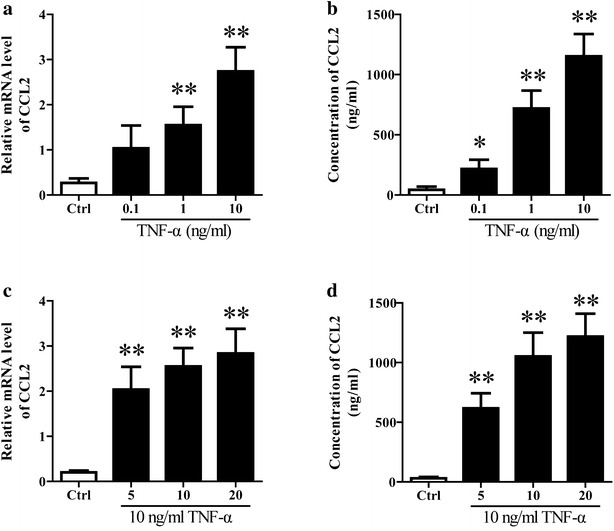
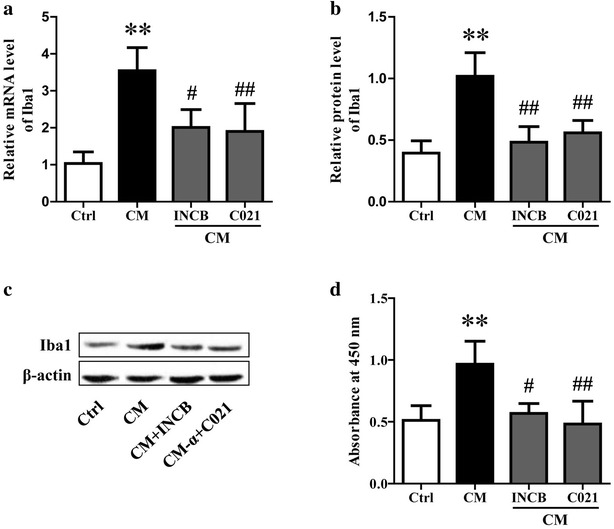
Methods: The rats were pretreated with CCL2 receptor inhibitors (INCB or C021, 1 mg/kg/day i.p.) for 3 days prior to thioacetamide (TAA) administration (300 mg/kg/day i.p.) for inducing HE model. At 8 h following the last injection (and every 4 h after), the grade of encephalopathy was assessed. Blood and whole brains were collected at coma for measuring CCL2 and Iba1 expression. In vitro, primary neurons were stimulated with TNF-α, and then the medium were collected for addition to microglia cultures with or without INCB or C021 pretreatment. The effect of the medium on microglia proliferation and activation was evaluated after 24 h.
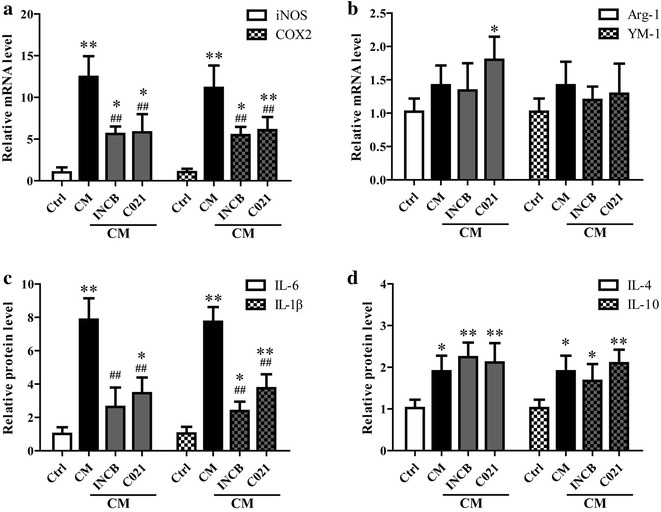
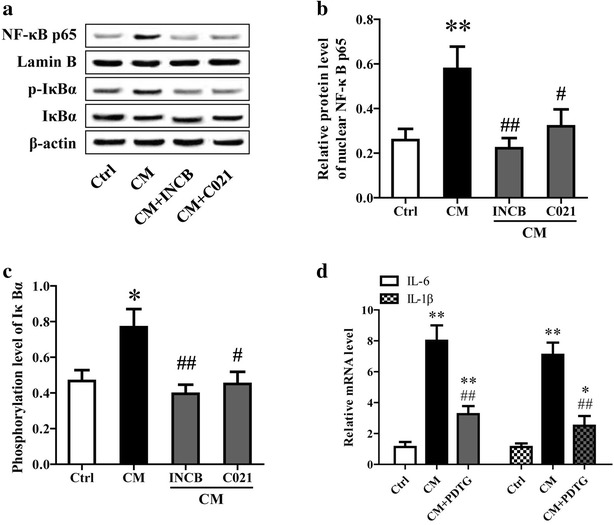
Results: CCL2 expression and microglia activation were elevated in the cerebral cortex of rats received TAA alone. CCL2 receptors inhibition improved neurological score and reduced cortical microglia activation. In vitro, TNF-α treatment induced CCL2 release by neurons. Medium from TNF-α stimulated neurons caused microglia proliferation and M1 markers expression, including iNOS, COX2, IL-6 and IL-1β, which could be suppressed by INCB or C021 pretreatment. The medium could also facilitate p65 nuclear translocation and IκBα phosphorylation, and NF-κB inhibition reduced the increased IL-6 and IL-1β expression induced by the medium.
Conclusion: Neuron-derived CCL2 contributed to microglia activation and neurological decline in HE. Blocking CCL2 or inhibiting microglia excessive activation may be potential strategies for HE.
Keywords: Chemokine CC motif ligand 2; Hepatic encephalopathy; Microglia; Neuron.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
