

Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia
- PMID: 28871148
- PMCID: PMC5583283
- DOI: 10.1038/s41467-017-00479-7
Editing an α-globin enhancer in primary human hematopoietic stem cells as a treatment for β-thalassemia
Abstract
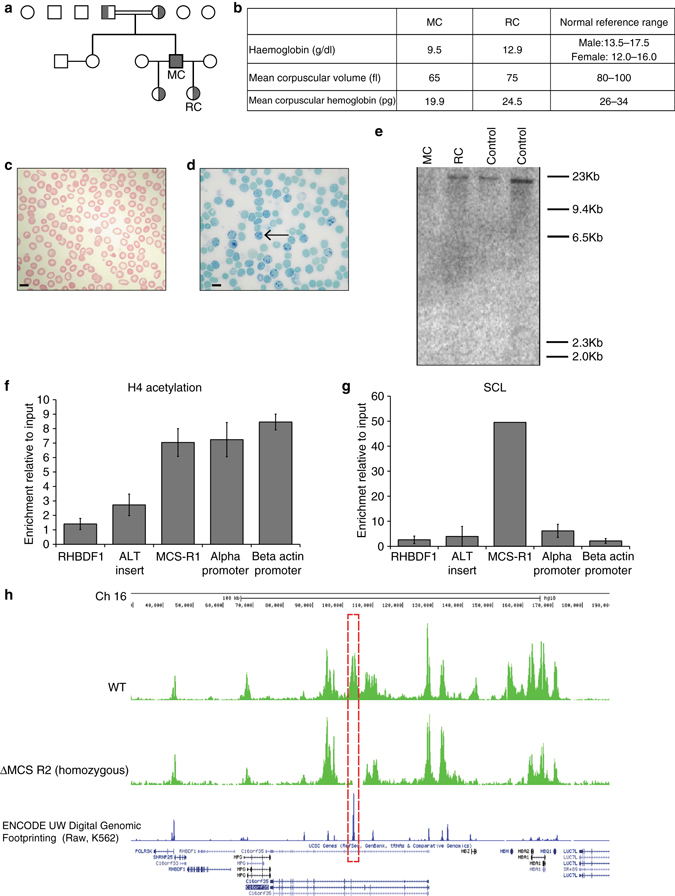
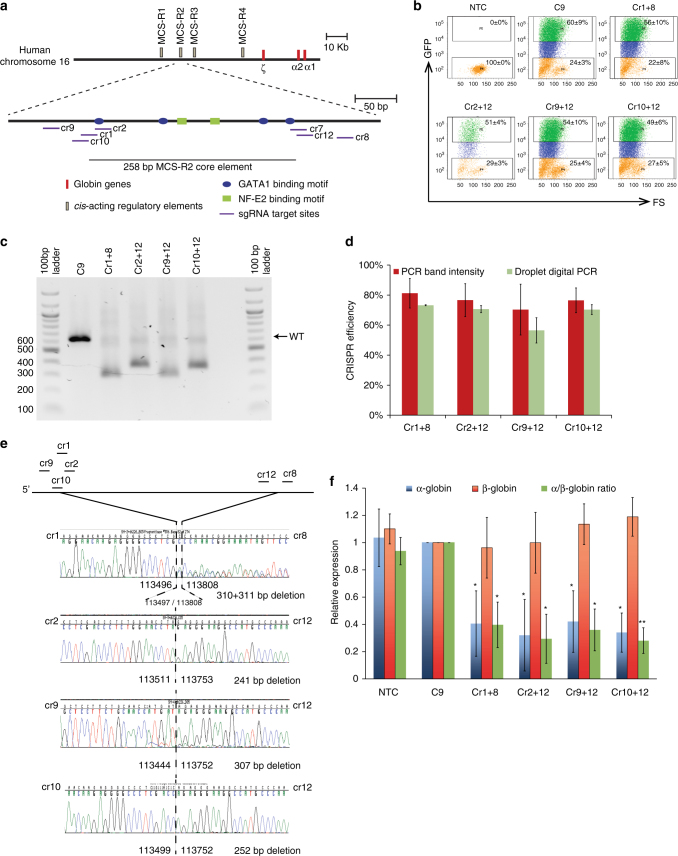
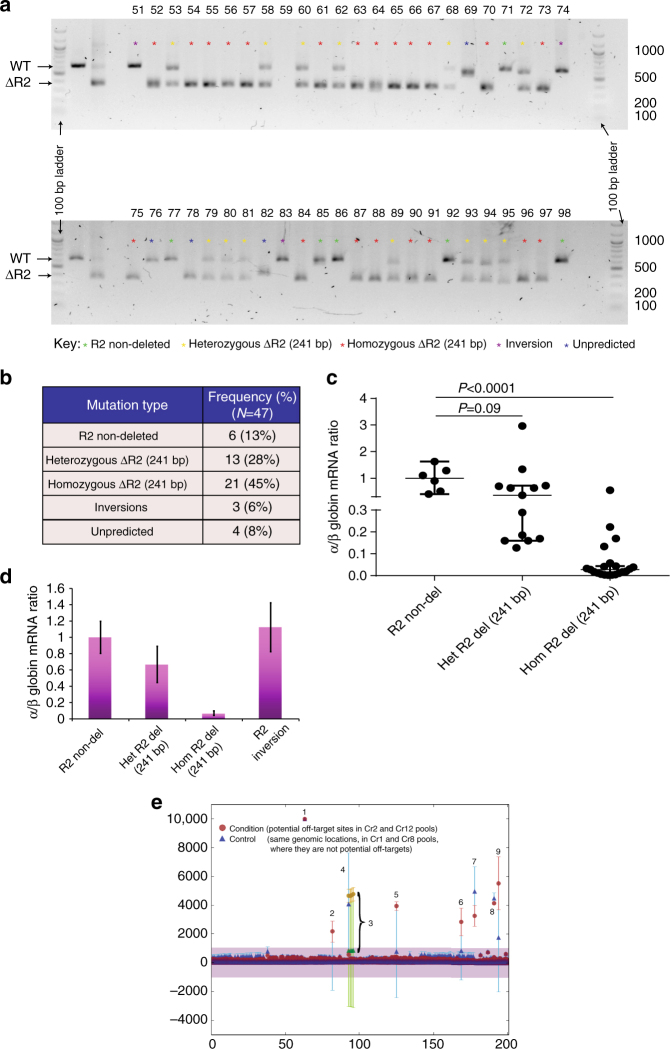
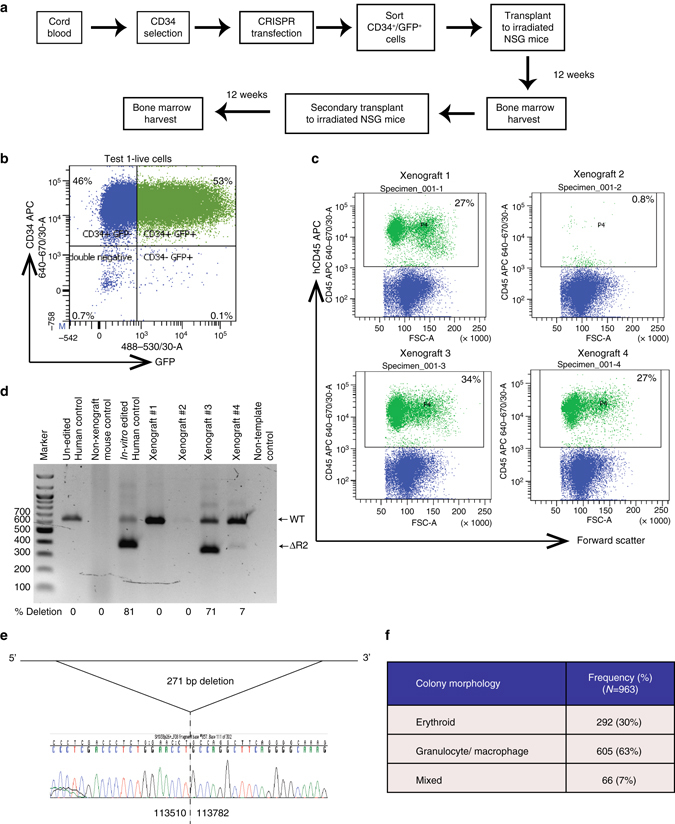
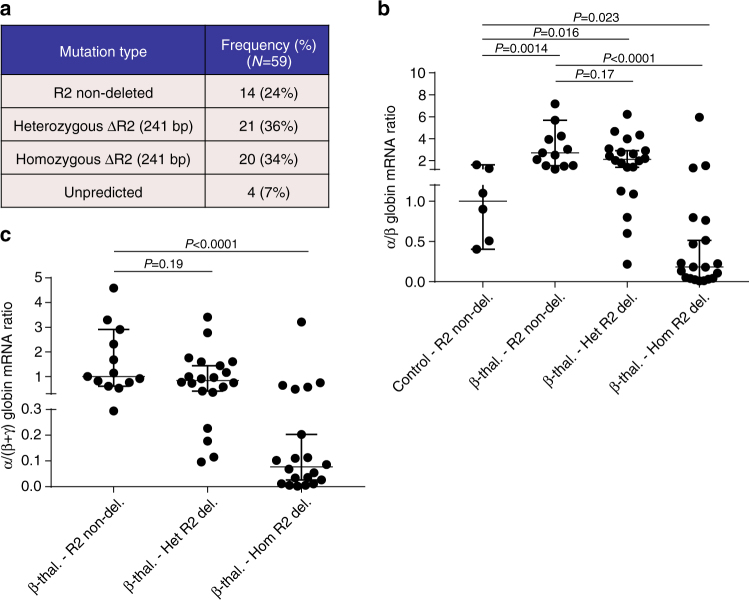
β-Thalassemia is one of the most common inherited anemias, with no effective cure for most patients. The pathophysiology reflects an imbalance between α- and β-globin chains with an excess of free α-globin chains causing ineffective erythropoiesis and hemolysis. When α-thalassemia is co-inherited with β-thalassemia, excess free α-globin chains are reduced significantly ameliorating the clinical severity. Here we demonstrate the use of CRISPR/Cas9 genome editing of primary human hematopoietic stem/progenitor (CD34+) cells to emulate a natural mutation, which deletes the MCS-R2 α-globin enhancer and causes α-thalassemia. When edited CD34+ cells are differentiated into erythroid cells, we observe the expected reduction in α-globin expression and a correction of the pathologic globin chain imbalance in cells from patients with β-thalassemia. Xenograft assays show that a proportion of the edited CD34+ cells are long-term repopulating hematopoietic stem cells, demonstrating the potential of this approach for translation into a therapy for β-thalassemia.β-thalassemia is characterised by the presence of an excess of α-globin chains, which contribute to erythrocyte pathology. Here the authors use CRISP/Cas9 to reduce α-globin expression in hematopoietic precursors, and show effectiveness in xenograft assays in mice.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Weatherall D. J. & Clegg J. B. in The Thalassaemia Syndromes, 4 edn (Blackwell Science, 2001).
Publication types
MeSH terms
Substances
Grants and funding
- MC_UU_12009/15/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/11/MRC_/Medical Research Council/United Kingdom
- MC_U137961145/MRC_/Medical Research Council/United Kingdom
- MR/L008963/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_12009/11/MRC_/Medical Research Council/United Kingdom
- MC_UU_12009/3/MRC_/Medical Research Council/United Kingdom
- MC_U137961144/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/14/MRC_/Medical Research Council/United Kingdom
- MC_PC_15069/MRC_/Medical Research Council/United Kingdom
- G1000801/MRC_/Medical Research Council/United Kingdom
- MC_UU_12009/4/MRC_/Medical Research Council/United Kingdom
- MR/P019633/1/MRC_/Medical Research Council/United Kingdom
- G1000729/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/3/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/4/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
