

mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy
- PMID: 28872463
- PMCID: PMC5617676
- DOI: 10.1172/JCI94130
mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy
Abstract
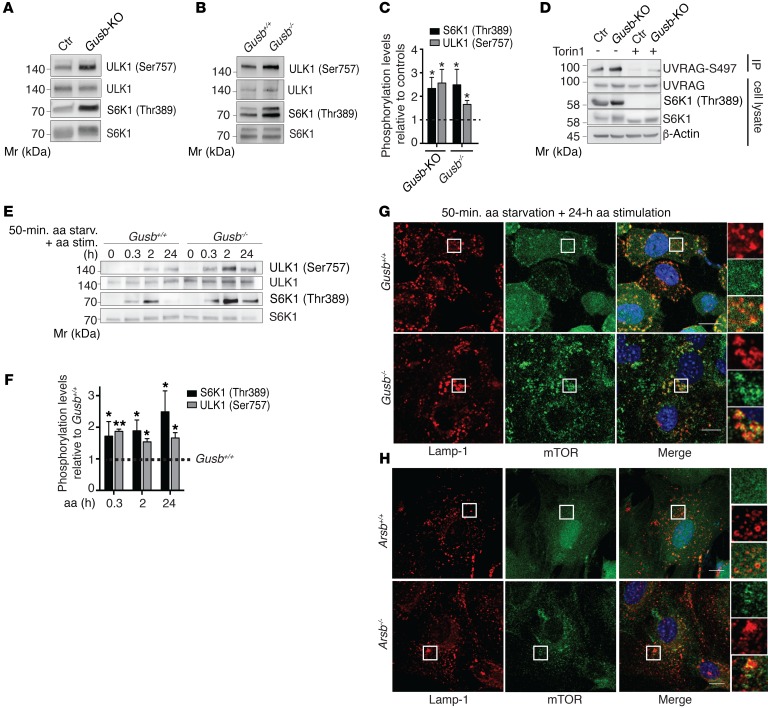
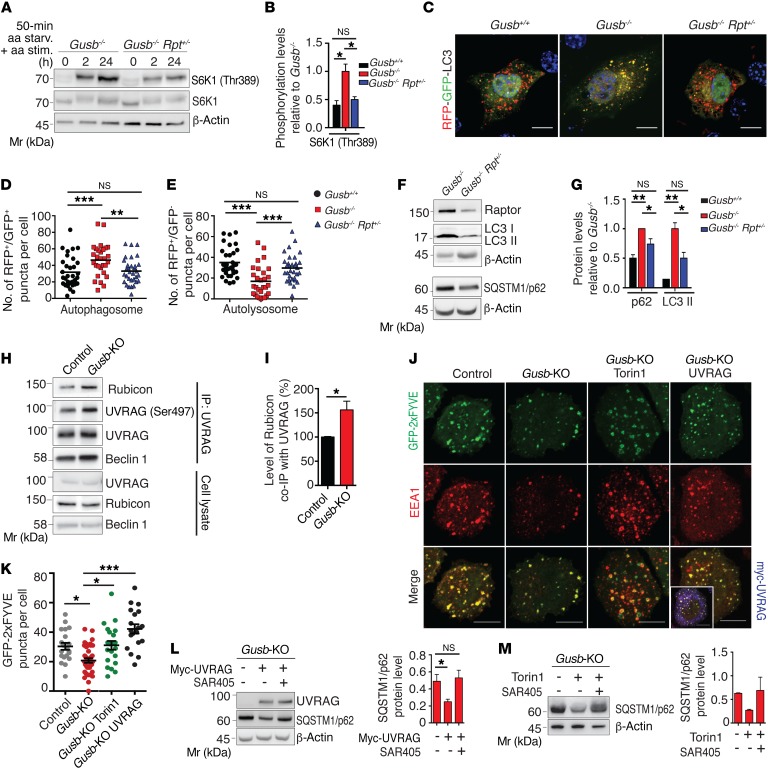
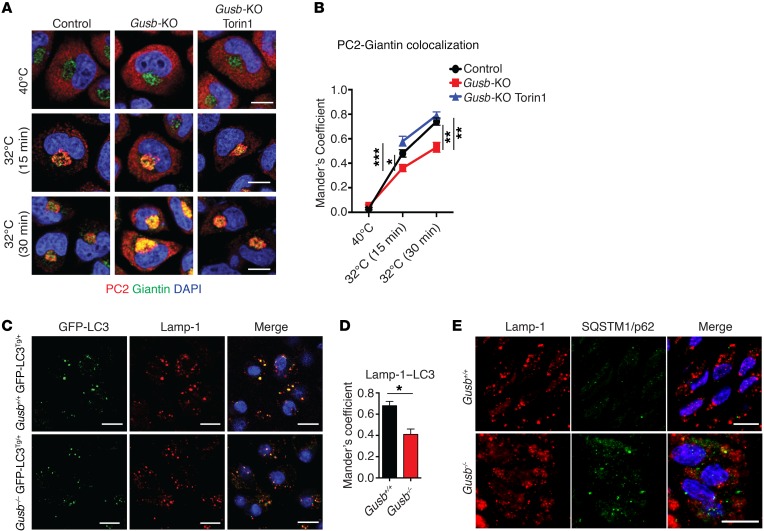
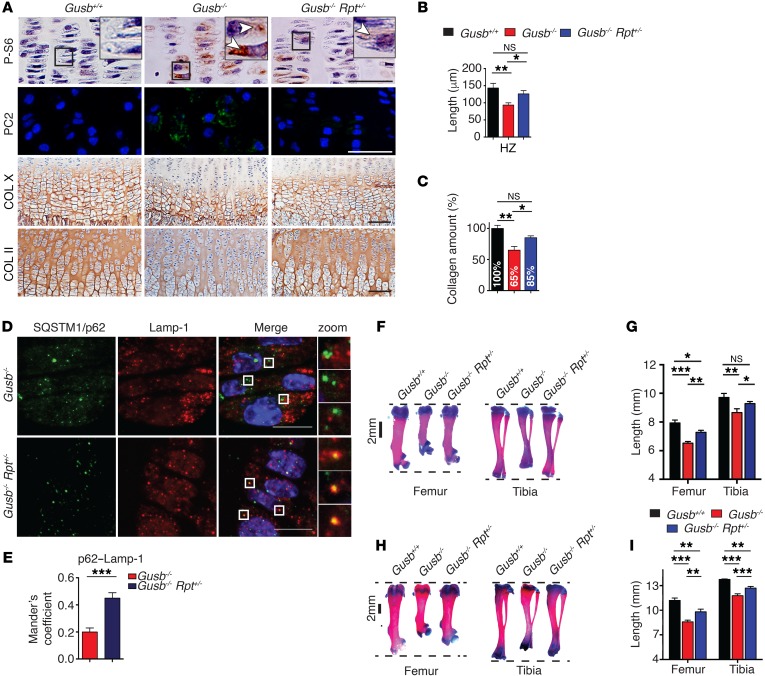
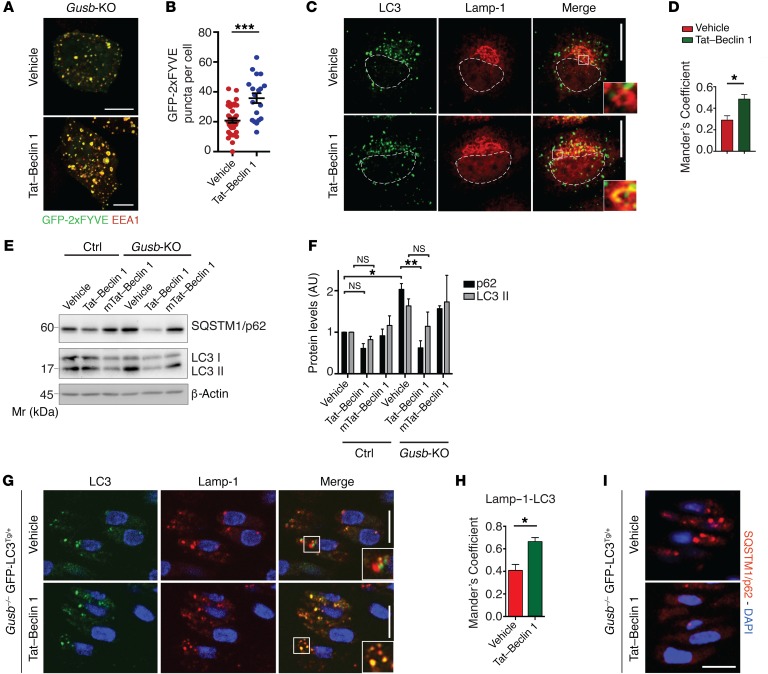
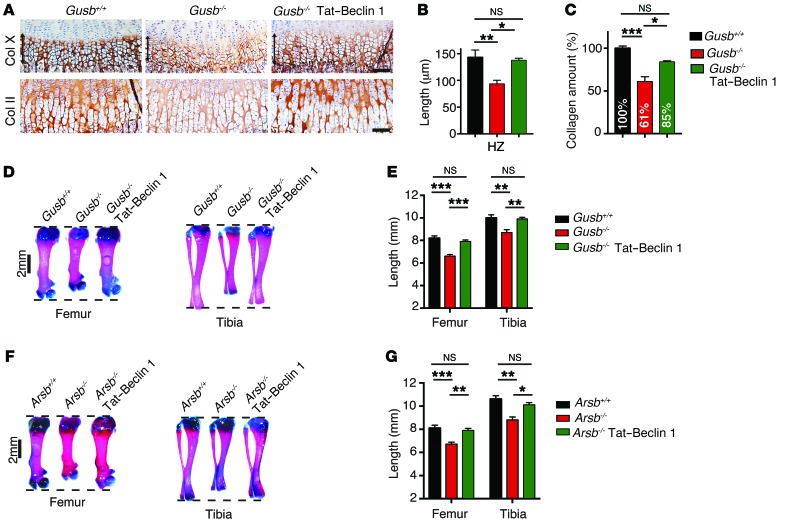
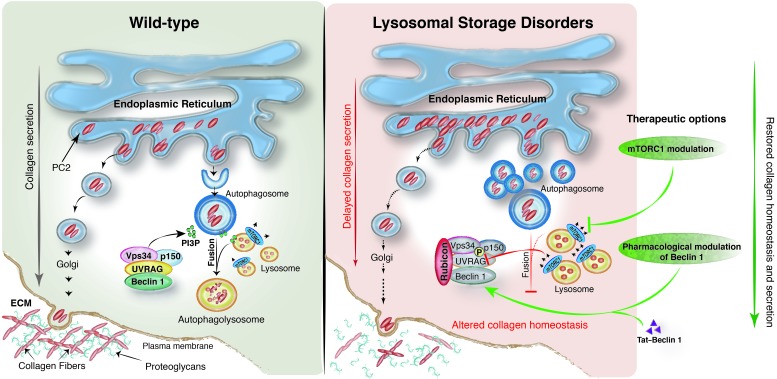
The mammalian target of rapamycin complex 1 (mTORC1) kinase promotes cell growth by activating biosynthetic pathways and suppressing catabolic pathways, particularly that of macroautophagy. A prerequisite for mTORC1 activation is its translocation to the lysosomal surface. Deregulation of mTORC1 has been associated with the pathogenesis of several diseases, but its role in skeletal disorders is largely unknown. Here, we show that enhanced mTORC1 signaling arrests bone growth in lysosomal storage disorders (LSDs). We found that lysosomal dysfunction induces a constitutive lysosomal association and consequent activation of mTORC1 in chondrocytes, the cells devoted to bone elongation. mTORC1 hyperphosphorylates the protein UV radiation resistance-associated gene (UVRAG), reducing the activity of the associated Beclin 1-Vps34 complex and thereby inhibiting phosphoinositide production. Limiting phosphoinositide production leads to a blockage of the autophagy flux in LSD chondrocytes. As a consequence, LSD chondrocytes fail to properly secrete collagens, the main components of the cartilage extracellular matrix. In mouse models of LSD, normalization of mTORC1 signaling or stimulation of the Beclin 1-Vps34-UVRAG complex rescued the autophagy flux, restored collagen levels in cartilage, and ameliorated the bone phenotype. Taken together, these data unveil a role for mTORC1 and autophagy in the pathogenesis of skeletal disorders and suggest potential therapeutic approaches for the treatment of LSDs.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
