

Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release
- PMID: 28874589
- PMCID: PMC5617277
- DOI: 10.1073/pnas.1705768114
Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release
Abstract
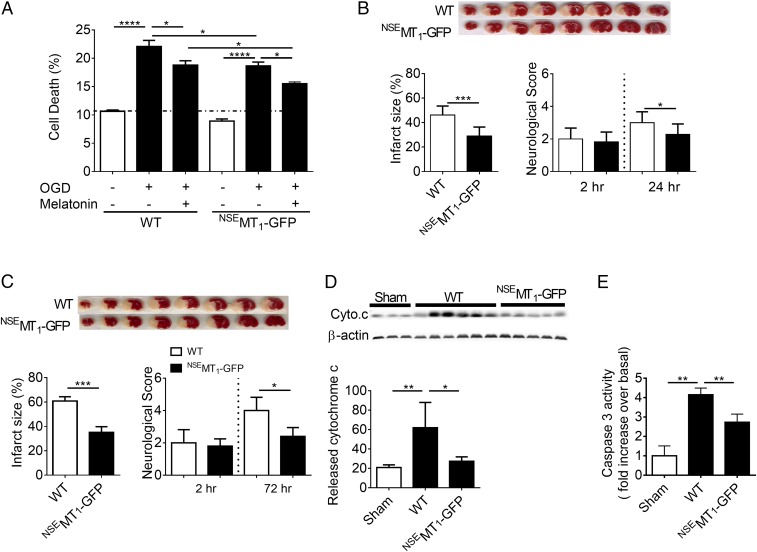
G protein-coupled receptors (GPCRs) are classically characterized as cell-surface receptors transmitting extracellular signals into cells. Here we show that central components of a GPCR signaling system comprised of the melatonin type 1 receptor (MT1), its associated G protein, and β-arrestins are on and within neuronal mitochondria. We discovered that the ligand melatonin is exclusively synthesized in the mitochondrial matrix and released by the organelle activating the mitochondrial MT1 signal-transduction pathway inhibiting stress-mediated cytochrome c release and caspase activation. These findings coupled with our observation that mitochondrial MT1 overexpression reduces ischemic brain injury in mice delineate a mitochondrial GPCR mechanism contributing to the neuroprotective action of melatonin. We propose a new term, "automitocrine," analogous to "autocrine" when a similar phenomenon occurs at the cellular level, to describe this unexpected intracellular organelle ligand-receptor pathway that opens a new research avenue investigating mitochondrial GPCR biology.
Keywords: G protein-coupled receptor; ischemia; melatonin; mitochondria; neuroprotection.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
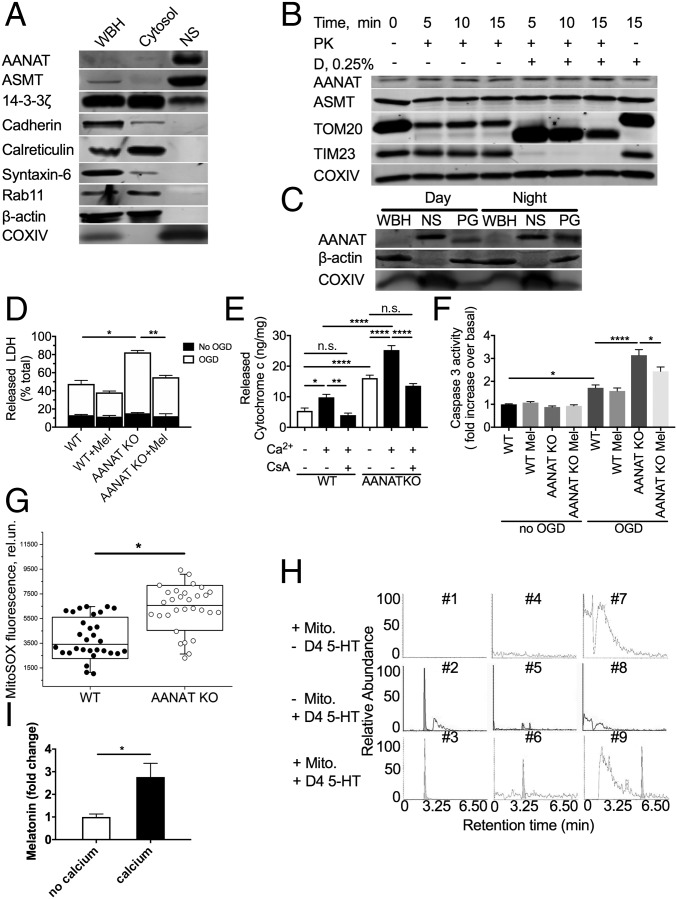
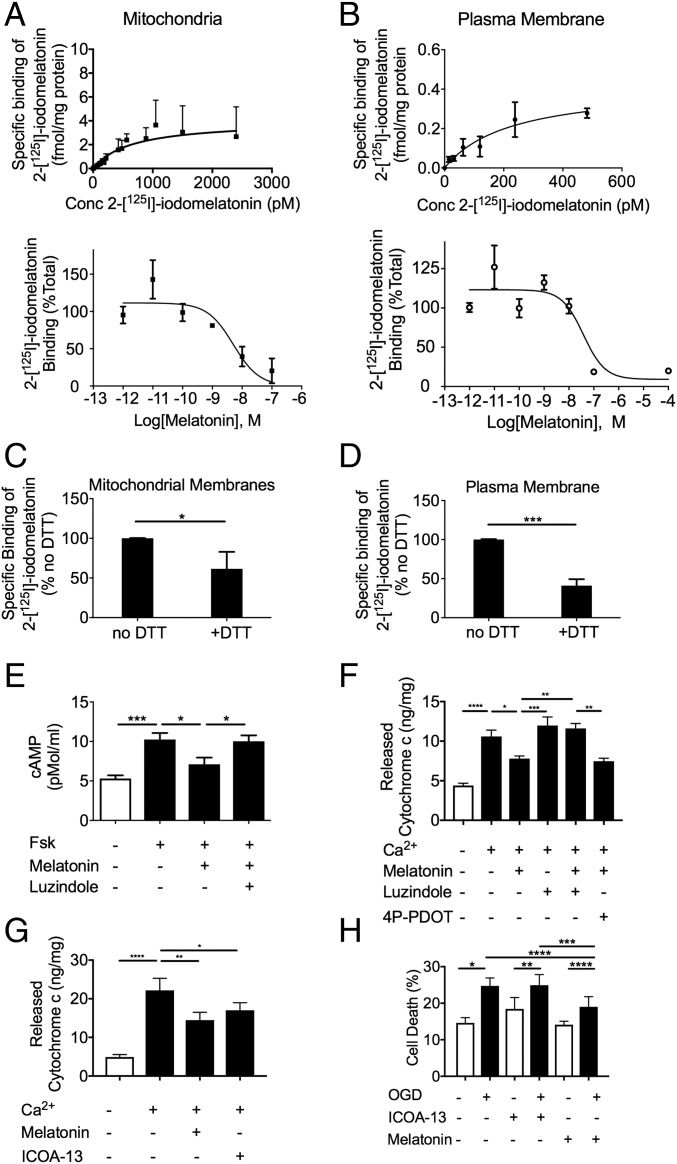
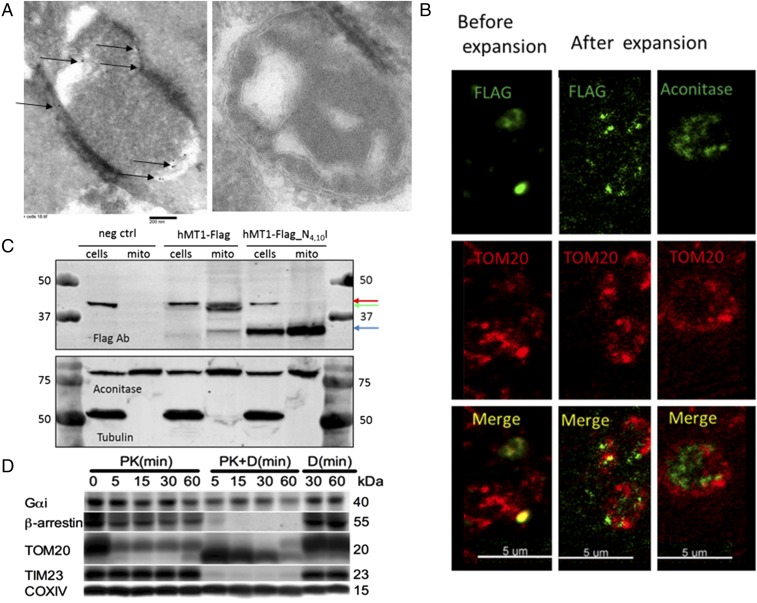
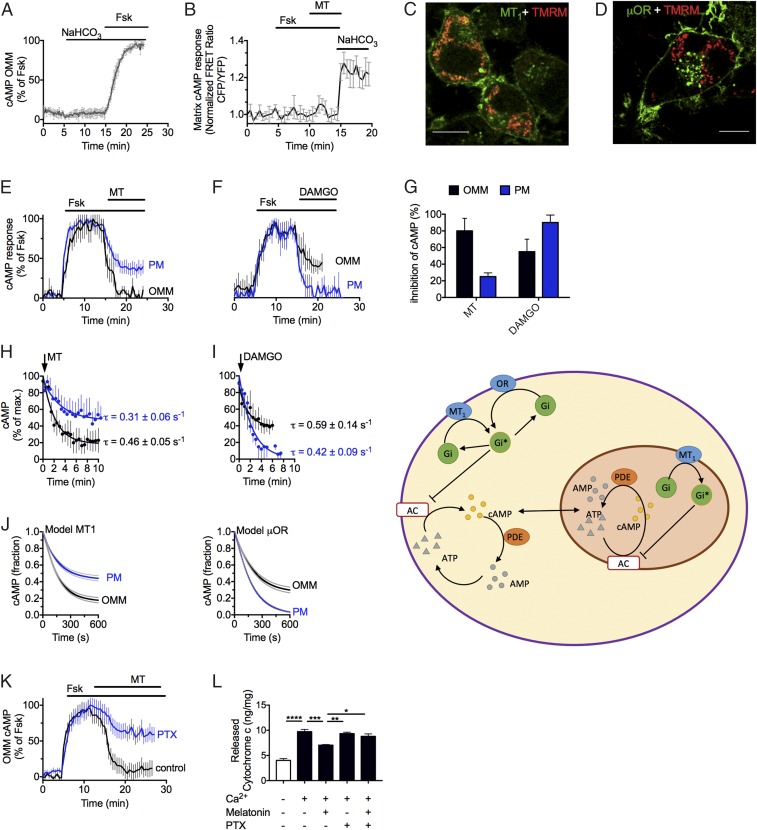
Comment in
-
Reply to Ahluwalia et al.: Contributions of melatonin receptors are tissue-dependent.Proc Natl Acad Sci U S A. 2018 Feb 27;115(9):E1944. doi: 10.1073/pnas.1800449115. Epub 2018 Feb 9. Proc Natl Acad Sci U S A. 2018. PMID: 29440383 Free PMC article. No abstract available.
-
Melatonin signaling in mitochondria extends beyond neurons and neuroprotection: Implications for angiogenesis and cardio/gastroprotection.Proc Natl Acad Sci U S A. 2018 Feb 27;115(9):E1942-E1943. doi: 10.1073/pnas.1722131115. Epub 2018 Feb 9. Proc Natl Acad Sci U S A. 2018. PMID: 29440384 Free PMC article. No abstract available.
References
-
- Poeggeler B, et al. Melatonin reverses the profibrillogenic activity of apolipoprotein E4 on the Alzheimer amyloid Abeta peptide. Biochemistry. 2001;40:14995–15001. - PubMed
-
- Reiter RJ, et al. Melatonin ameliorates neurologic damage and neurophysiologic deficits in experimental models of stroke. Ann N Y Acad Sci. 2003;993:35–47, discussion 48–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
