

Phenotype-driven precision oncology as a guide for clinical decisions one patient at a time
- PMID: 28874669
- PMCID: PMC5585361
- DOI: 10.1038/s41467-017-00451-5
Phenotype-driven precision oncology as a guide for clinical decisions one patient at a time
Abstract
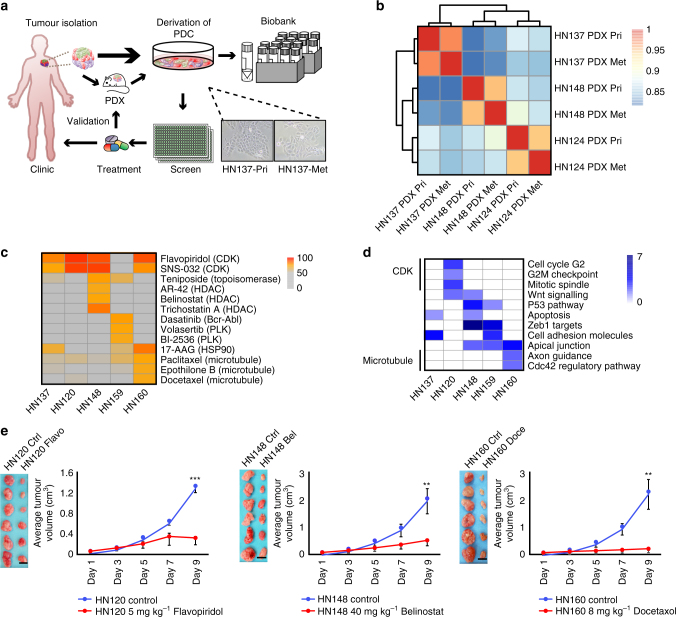
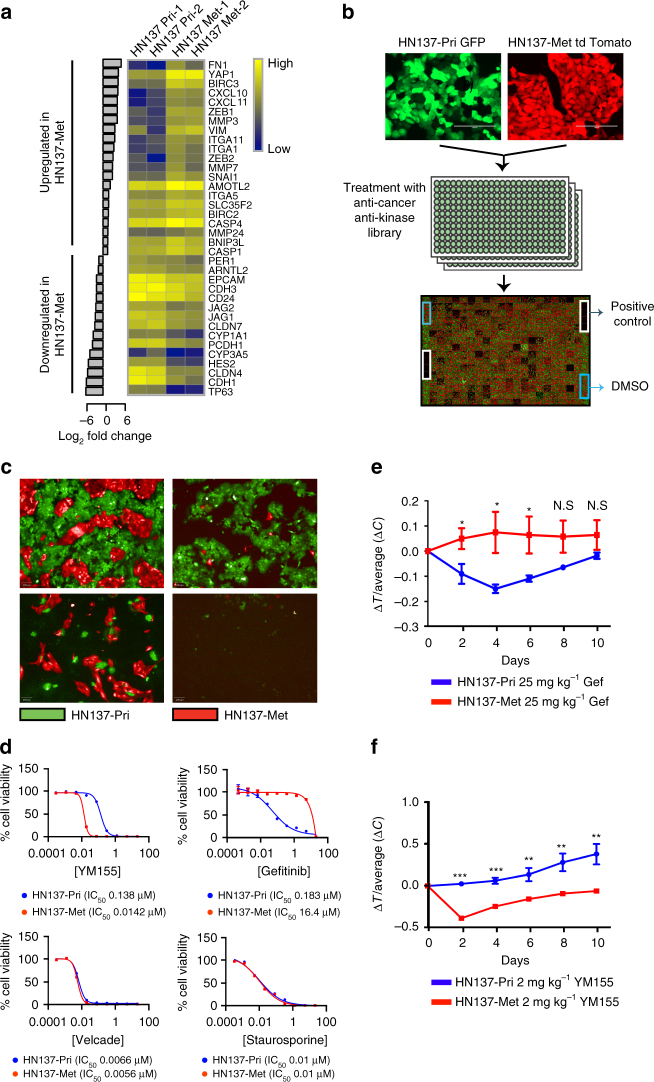
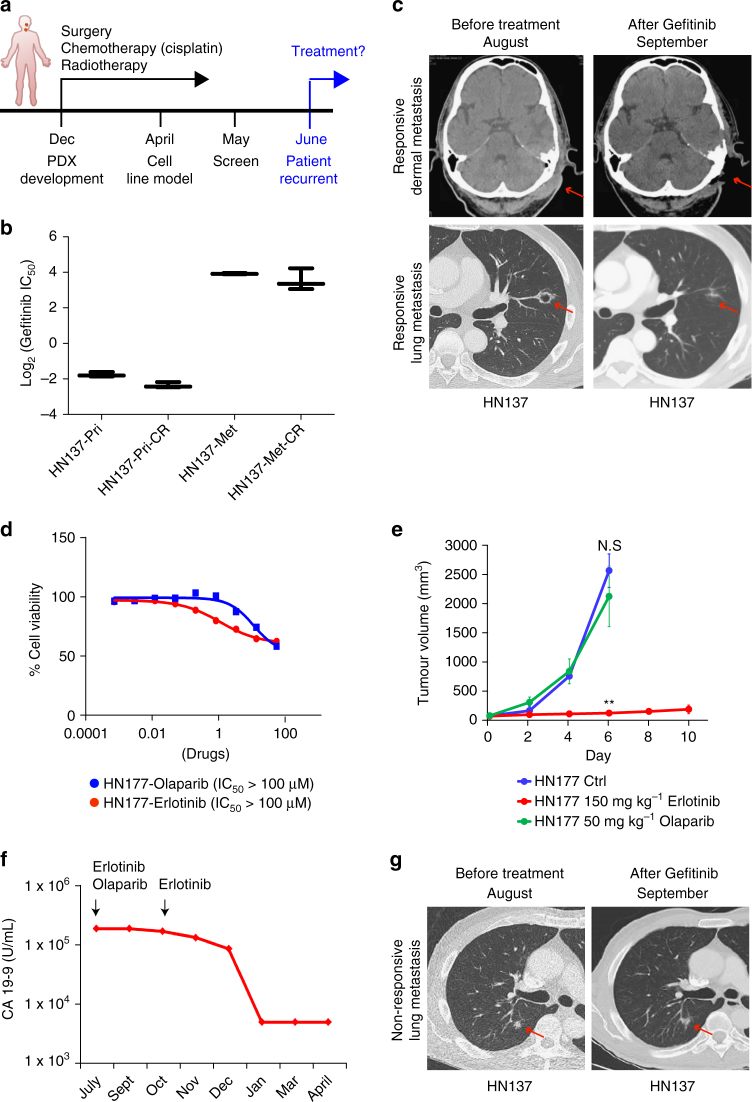
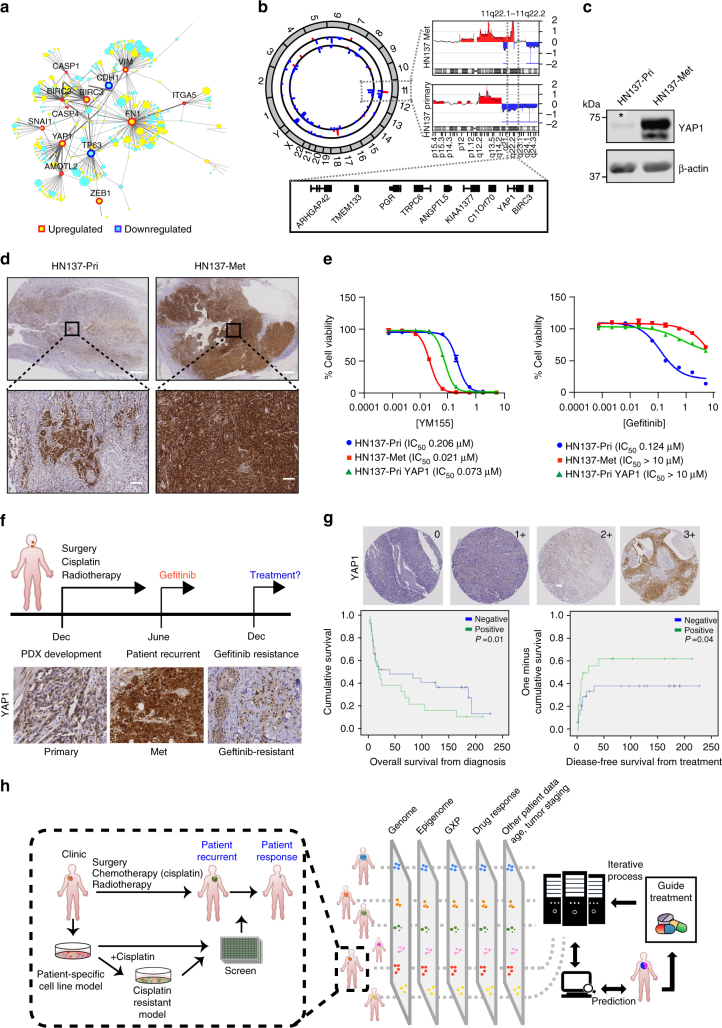
Genomics-driven cancer therapeutics has gained prominence in personalized cancer treatment. However, its utility in indications lacking biomarker-driven treatment strategies remains limited. Here we present a "phenotype-driven precision-oncology" approach, based on the notion that biological response to perturbations, chemical or genetic, in ex vivo patient-individualized models can serve as predictive biomarkers for therapeutic response in the clinic. We generated a library of "screenable" patient-derived primary cultures (PDCs) for head and neck squamous cell carcinomas that reproducibly predicted treatment response in matched patient-derived-xenograft models. Importantly, PDCs could guide clinical practice and predict tumour progression in two n = 1 co-clinical trials. Comprehensive "-omics" interrogation of PDCs derived from one of these models revealed YAP1 as a putative biomarker for treatment response and survival in ~24% of oral squamous cell carcinoma. We envision that scaling of the proposed PDC approach could uncover biomarkers for therapeutic stratification and guide real-time therapeutic decisions in the future.Treatment response in patient-derived models may serve as a biomarker for response in the clinic. Here, the authors use paired patient-derived mouse xenografts and patient-derived primary culture models from head and neck squamous cell carcinomas, including metastasis, as models for high-throughput screening of anti-cancer drugs.
Trial registration: ClinicalTrials.gov NCT02806388.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
