

Surveillance for highly pathogenic influenza A viruses in California during 2014-2015 provides insights into viral evolutionary pathways and the spatiotemporal extent of viruses in the Pacific Americas Flyway
- PMID: 28874792
- PMCID: PMC5625317
- DOI: 10.1038/emi.2017.66
Surveillance for highly pathogenic influenza A viruses in California during 2014-2015 provides insights into viral evolutionary pathways and the spatiotemporal extent of viruses in the Pacific Americas Flyway
Abstract
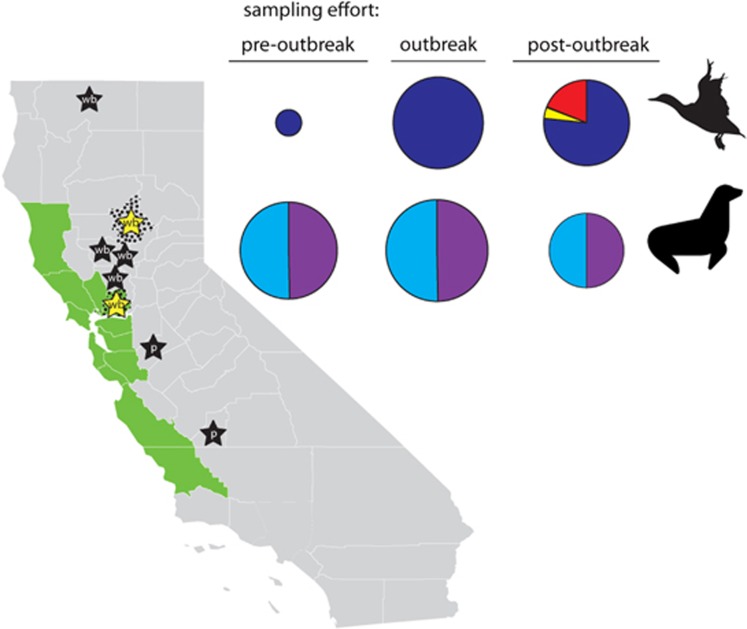
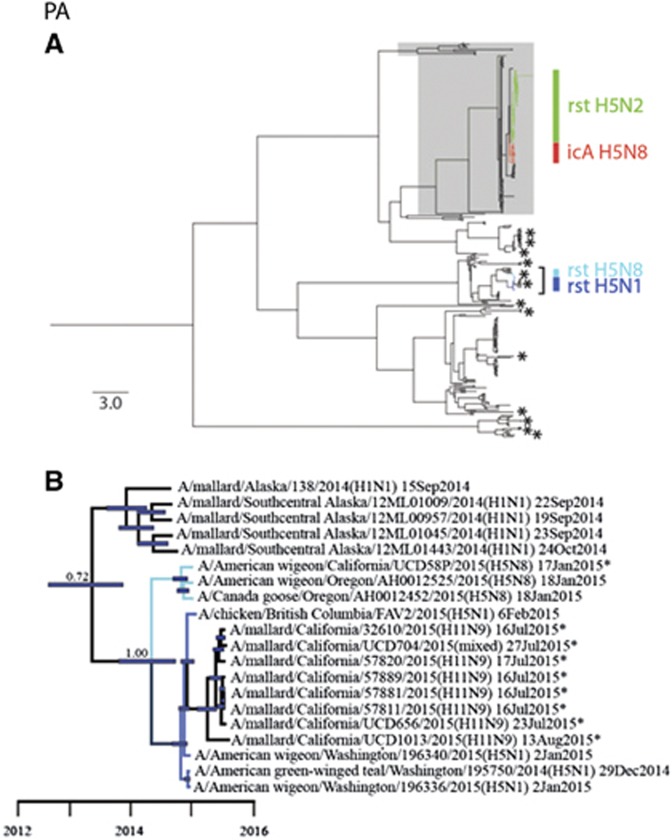
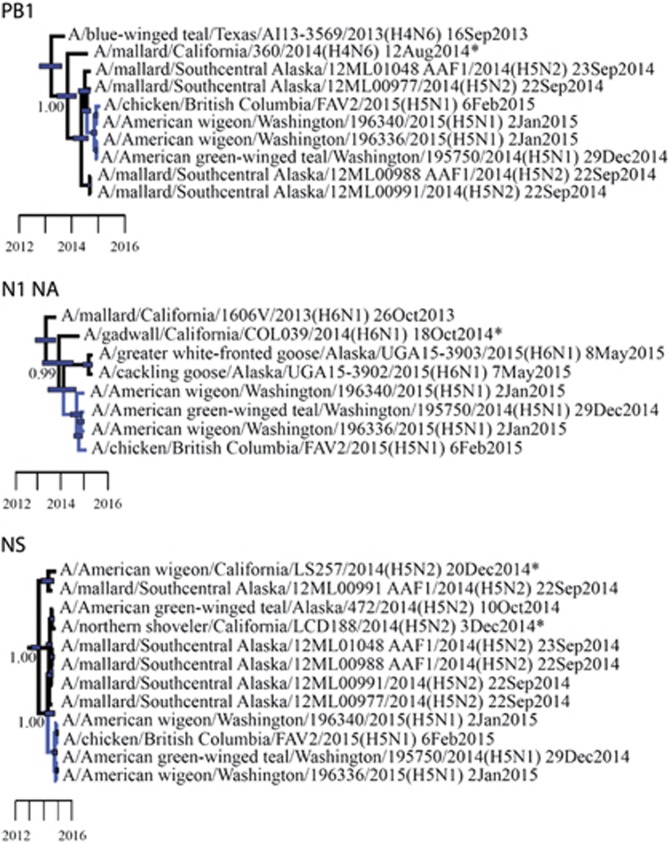
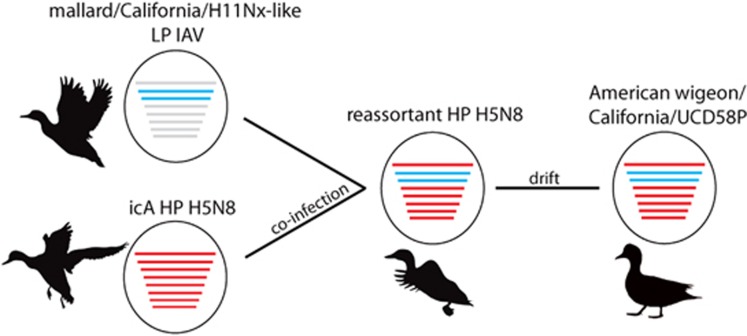
We used surveillance data collected in California before, concurrent with, and subsequent to an outbreak of highly pathogenic (HP) clade 2.3.4.4 influenza A viruses (IAVs) in 2014-2015 to (i) evaluate IAV prevalence in waterfowl, (ii) assess the evidence for spill-over infections in marine mammals and (iii) genetically characterize low-pathogenic (LP) and HP IAVs to refine inference on the spatiotemporal extent of HP genome constellations and to evaluate possible evolutionary pathways. We screened samples from 1496 waterfowl and 1142 marine mammals collected from April 2014 to August 2015 and detected IAV RNA in 159 samples collected from birds (n=157) and pinnipeds (n=2). HP IAV RNA was identified in three samples originating from American wigeon (Anas americana). Genetic sequence data were generated for a clade 2.3.4.4 HP IAV-positive diagnostic sample and 57 LP IAV isolates. Phylogenetic analyses revealed that the HP IAV was a reassortant H5N8 virus with gene segments closely related to LP IAVs detected in mallards (Anas platyrhynchos) sampled in California and other IAVs detected in wild birds sampled within the Pacific Americas Flyway. In addition, our analysis provided support for common ancestry between LP IAVs recovered from waterfowl sampled in California and gene segments of reassortant HP H5N1 IAVs detected in British Columbia, Canada and Washington, USA. Our investigation provides evidence that waterfowl are likely to have played a role in the evolution of reassortant HP IAVs in the Pacific Americas Flyway during 2014-2015, whereas we did not find support for spill-over infections in potential pinniped hosts.
Figures

References
-
- Li KS, Guan Y, Wang J et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 2004; 430: 209–213. - PubMed
-
- Zhao K, Gu M, Zhong L et al. Characterization of three H5N5 and one H5N8 highly pathogenic avian influenza viruses in China. Vet Microbiol 2013; 163: 351–357. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous