

The Schistosoma mansoni genome encodes thousands of long non-coding RNAs predicted to be functional at different parasite life-cycle stages
- PMID: 28874839
- PMCID: PMC5585378
- DOI: 10.1038/s41598-017-10853-6
The Schistosoma mansoni genome encodes thousands of long non-coding RNAs predicted to be functional at different parasite life-cycle stages
Abstract
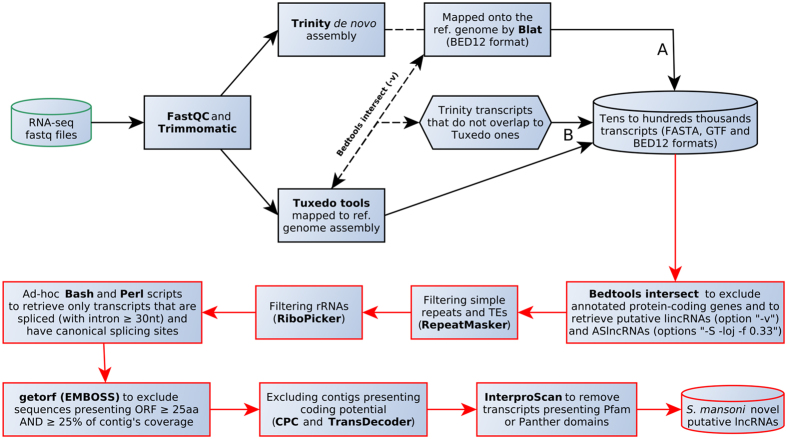
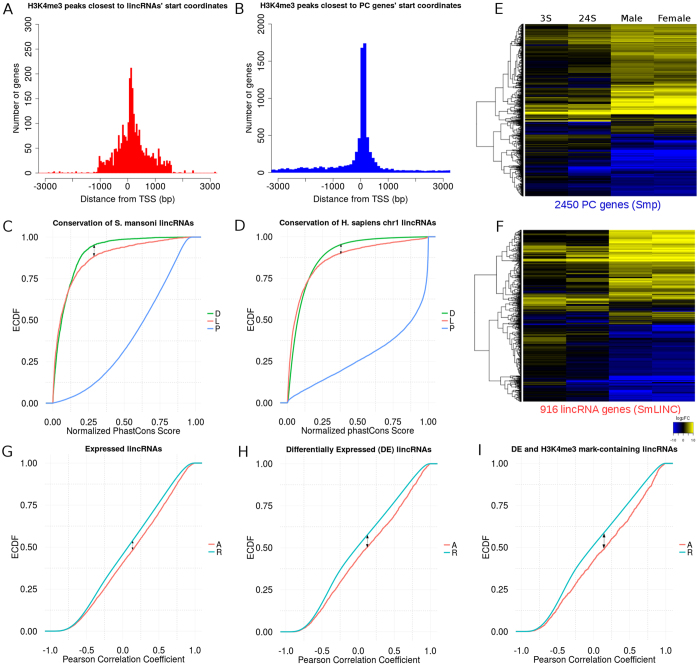
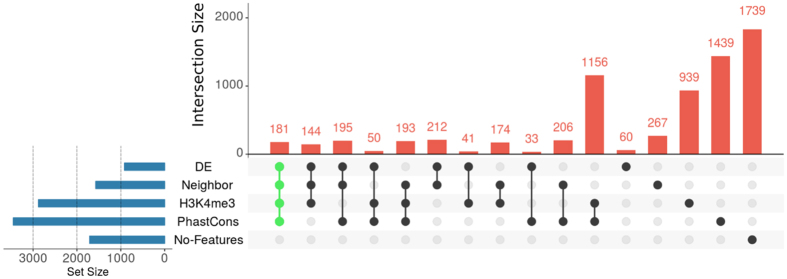
Next Generation Sequencing (NGS) strategies, like RNA-Seq, have revealed the transcription of a wide variety of long non-coding RNAs (lncRNAs) in the genomes of several organisms. In the present work we assessed the lncRNAs complement of Schistosoma mansoni, the blood fluke that causes schistosomiasis, ranked among the most prevalent parasitic diseases worldwide. We focused on the long intergenic/intervening ncRNAs (lincRNAs), hidden within the large amount of information obtained through RNA-Seq in S. mansoni (88 libraries). Our computational pipeline identified 7029 canonically-spliced putative lincRNA genes on 2596 genomic loci (at an average 2.7 isoforms per lincRNA locus), as well as 402 spliced lncRNAs that are antisense to protein-coding (PC) genes. Hundreds of lincRNAs showed traits for being functional, such as the presence of epigenetic marks at their transcription start sites, evolutionary conservation among other schistosome species and differential expression across five different life-cycle stages of the parasite. Real-time qPCR has confirmed the differential life-cycle stage expression of a set of selected lincRNAs. We have built PC gene and lincRNA co-expression networks, unraveling key biological processes where lincRNAs might be involved during parasite development. This is the first report of a large-scale identification and structural annotation of lncRNAs in the S. mansoni genome.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




Similar articles
-
Atlas of Schistosoma mansoni long non-coding RNAs and their expression correlation to protein-coding genes.Database (Oxford). 2018 Jan 1;2018:bay068. doi: 10.1093/database/bay068. Database (Oxford). 2018. PMID: 29992321 Free PMC article.
-
Schistosoma mansoni Egg, Adult Male and Female Comparative Gene Expression Analysis and Identification of Novel Genes by RNA-Seq.PLoS Negl Trop Dis. 2015 Dec 31;9(12):e0004334. doi: 10.1371/journal.pntd.0004334. eCollection 2015 Dec. PLoS Negl Trop Dis. 2015. PMID: 26719891 Free PMC article.
-
Identification of long noncoding RNAs in Schistosoma mansoni and Schistosoma japonicum.Exp Parasitol. 2018 Aug;191:82-87. doi: 10.1016/j.exppara.2018.07.001. Epub 2018 Jul 4. Exp Parasitol. 2018. PMID: 29981293
-
Schistosoma mansoni genome project: an update.Parasitol Int. 2004 Jun;53(2):183-92. doi: 10.1016/j.parint.2004.01.009. Parasitol Int. 2004. PMID: 15081950 Review.
-
Non-coding RNAs in schistosomes: an unexplored world.An Acad Bras Cienc. 2011 Jun;83(2):673-94. doi: 10.1590/s0001-37652011000200026. An Acad Bras Cienc. 2011. PMID: 21670887 Review.
Cited by
-
Epigenetic Markers Associated with Schistosomiasis.Helminthologia. 2021 Feb 10;58(1):28-40. doi: 10.2478/helm-2021-0009. eCollection 2021 Mar. Helminthologia. 2021. PMID: 33664616 Free PMC article.
-
Identification of 170 New Long Noncoding RNAs in Schistosoma mansoni.Biomed Res Int. 2018 Jul 11;2018:1264697. doi: 10.1155/2018/1264697. eCollection 2018. Biomed Res Int. 2018. PMID: 30112357 Free PMC article.
-
Atlas of Schistosoma mansoni long non-coding RNAs and their expression correlation to protein-coding genes.Database (Oxford). 2018 Jan 1;2018:bay068. doi: 10.1093/database/bay068. Database (Oxford). 2018. PMID: 29992321 Free PMC article.
-
RNA sequencing analysis of altered expression of long noncoding RNAs associated with Schistosoma japonicum infection in the murine liver and spleen.Parasit Vectors. 2020 Dec 1;13(1):601. doi: 10.1186/s13071-020-04457-9. Parasit Vectors. 2020. PMID: 33261628 Free PMC article.
-
In Silico Analysis of Common Long Noncoding RNAs in Schistosoma mansoni and Schistosoma haematobium.J Trop Med. 2021 Feb 15;2021:6617118. doi: 10.1155/2021/6617118. eCollection 2021. J Trop Med. 2021. PMID: 33628277 Free PMC article.
References
-
- Mattick, J. S. & Makunin, I. V. Non-coding RNA. Hum Mol Genet15 Spec No 1, R17–29 (2006). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
