

Aging: Molecular Pathways and Implications on the Cardiovascular System
- PMID: 28874954
- PMCID: PMC5569936
- DOI: 10.1155/2017/7941563
Aging: Molecular Pathways and Implications on the Cardiovascular System
Abstract
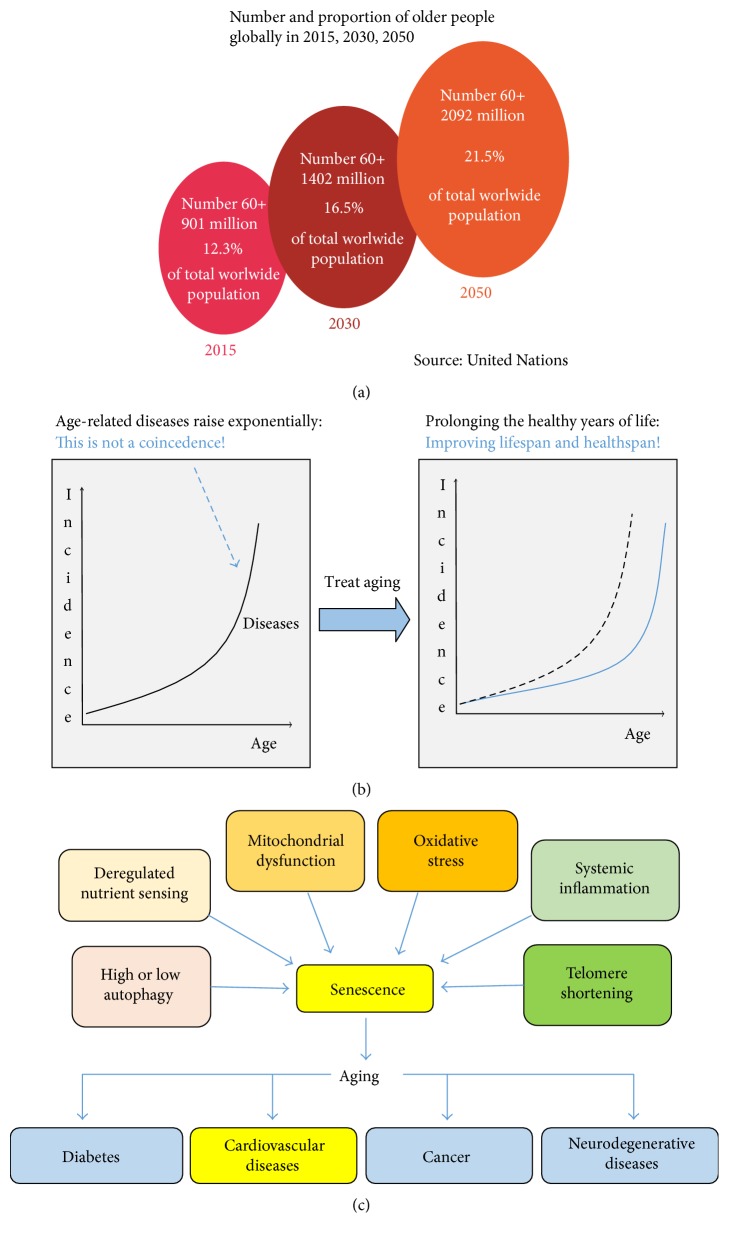
The world's population over 60 years is growing rapidly, reaching 22% of the global population in the next decades. Despite the increase in global longevity, individual healthspan needs to follow this growth. Several diseases have their prevalence increased by age, such as cardiovascular diseases, the leading cause of morbidity and mortality worldwide. Understanding the aging biology mechanisms is fundamental to the pursuit of cardiovascular health. In this way, aging is characterized by a gradual decline in physiological functions, involving the increased number in senescent cells into the body. Several pathways lead to senescence, including oxidative stress and persistent inflammation, as well as energy failure such as mitochondrial dysfunction and deregulated autophagy, being ROS, AMPK, SIRTs, mTOR, IGF-1, and p53 key regulators of the metabolic control, connecting aging to the pathways which drive towards diseases. In addition, senescence can be induced by cellular replication, which resulted from telomere shortening. Taken together, it is possible to draw a common pathway unifying aging to cardiovascular diseases, and the central point of this process, senescence, can be the target for new therapies, which may result in the healthspan matching the lifespan.
Figures







Similar articles
-
Theories and Molecular Basis of Vascular Aging: A Review of the Literature from VascAgeNet Group on Pathophysiological Mechanisms of Vascular Aging.Int J Mol Sci. 2022 Aug 4;23(15):8672. doi: 10.3390/ijms23158672. Int J Mol Sci. 2022. PMID: 35955804 Free PMC article. Review.
-
Molecular and cellular pathways contributing to brain aging.Behav Brain Funct. 2021 Jun 12;17(1):6. doi: 10.1186/s12993-021-00179-9. Behav Brain Funct. 2021. PMID: 34118939 Free PMC article. Review.
-
New Horizons: Novel Approaches to Enhance Healthspan Through Targeting Cellular Senescence and Related Aging Mechanisms.J Clin Endocrinol Metab. 2021 Mar 8;106(3):e1481-e1487. doi: 10.1210/clinem/dgaa728. J Clin Endocrinol Metab. 2021. PMID: 33155651 Free PMC article. Review.
-
Cellular and molecular biology of aging endothelial cells.J Mol Cell Cardiol. 2015 Dec;89(Pt B):122-35. doi: 10.1016/j.yjmcc.2015.01.021. Epub 2015 Feb 2. J Mol Cell Cardiol. 2015. PMID: 25655936 Free PMC article. Review.
-
Telomere and its role in the aging pathways: telomere shortening, cell senescence and mitochondria dysfunction.Biogerontology. 2019 Feb;20(1):1-16. doi: 10.1007/s10522-018-9769-1. Epub 2018 Sep 18. Biogerontology. 2019. PMID: 30229407 Review.
Cited by
-
Aging-related genes are potential prognostic biomarkers for patients with gliomas.Aging (Albany NY). 2021 May 4;13(9):13239-13263. doi: 10.18632/aging.203008. Epub 2021 May 4. Aging (Albany NY). 2021. PMID: 33946049 Free PMC article.
-
Biomedical generative pre-trained based transformer language model for age-related disease target discovery.Aging (Albany NY). 2023 Sep 22;15(18):9293-9309. doi: 10.18632/aging.205055. Epub 2023 Sep 22. Aging (Albany NY). 2023. PMID: 37742294 Free PMC article.
-
Metformin prevents age-associated ovarian fibrosis by modulating the immune landscape in female mice.Sci Adv. 2022 Sep 2;8(35):eabq1475. doi: 10.1126/sciadv.abq1475. Epub 2022 Sep 2. Sci Adv. 2022. PMID: 36054356 Free PMC article.
-
Management of Very Old Patients in Intensive Care Units.Aging Dis. 2021 Apr 1;12(2):614-624. doi: 10.14336/AD.2020.0914. eCollection 2021 Apr. Aging Dis. 2021. PMID: 33815886 Free PMC article. Review.
-
Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events.Front Physiol. 2018 Jul 3;9:806. doi: 10.3389/fphys.2018.00806. eCollection 2018. Front Physiol. 2018. PMID: 30018565 Free PMC article. Review.
References
-
- UN. HelpAge, Global AgeWatch Index 2015: Insight Report. London WC1A 9GB, UK: HelpAge International; 2015. UNDESA Population Division, World population prospects: the 2015 revision.
-
- WHO. NCD Mortality and Morbidity. World Health Organization; 2012. http://www.who.int/gho/ncd/mortality_morbidity/en/
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
