

Atropos: specific, sensitive, and speedy trimming of sequencing reads
- PMID: 28875074
- PMCID: PMC5581536
- DOI: 10.7717/peerj.3720
Atropos: specific, sensitive, and speedy trimming of sequencing reads
Abstract
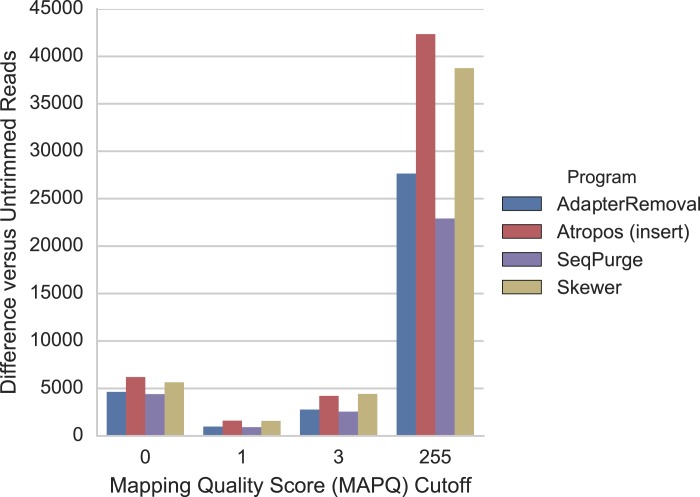
A key step in the transformation of raw sequencing reads into biological insights is the trimming of adapter sequences and low-quality bases. Read trimming has been shown to increase the quality and reliability while decreasing the computational requirements of downstream analyses. Many read trimming software tools are available; however, no tool simultaneously provides the accuracy, computational efficiency, and feature set required to handle the types and volumes of data generated in modern sequencing-based experiments. Here we introduce Atropos and show that it trims reads with high sensitivity and specificity while maintaining leading-edge speed. Compared to other state-of-the-art read trimming tools, Atropos achieves significant increases in trimming accuracy while remaining competitive in execution times. Furthermore, Atropos maintains high accuracy even when trimming data with elevated rates of sequencing errors. The accuracy, high performance, and broad feature set offered by Atropos makes it an appropriate choice for the pre-processing of Illumina, ABI SOLiD, and other current-generation short-read sequencing datasets. Atropos is open source and free software written in Python (3.3+) and available at https://github.com/jdidion/atropos.
Keywords: Adapter; Cutadapt; Illumina; NGS; Preprocessing; Read; Sequencing; Trimming.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
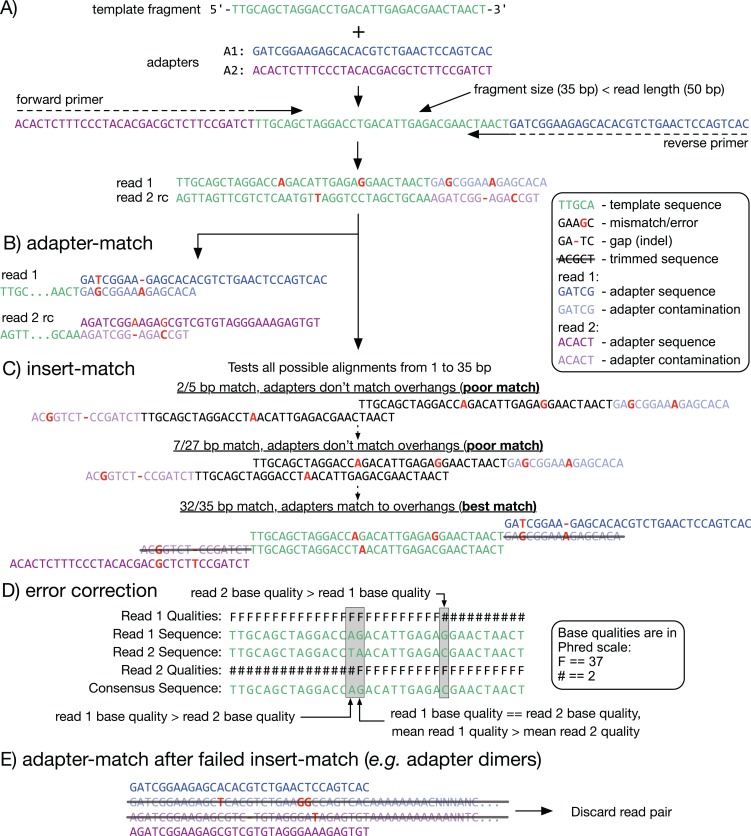
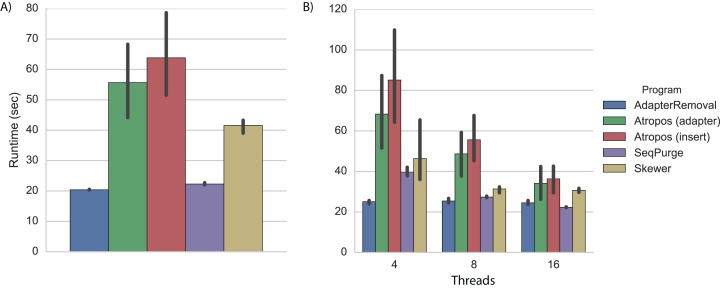
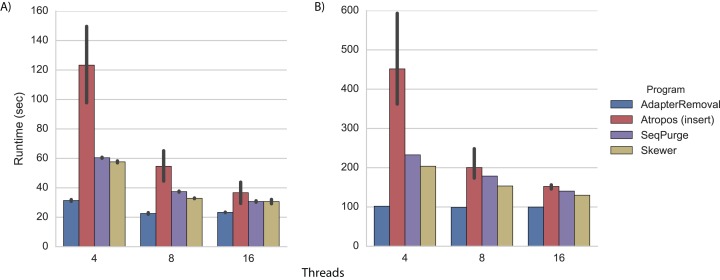
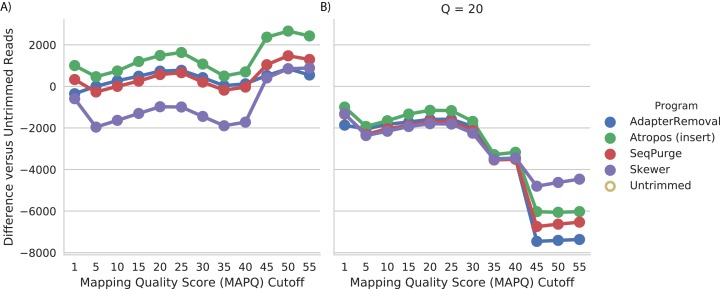
References
-
- Andrews S. FastQC: a quality control tool for high throughput sequence data. Version: 0.11.5http://www.bioinformatics.babraham.ac.uk/projects/fastqc 2010
-
- Boettiger C. An introduction to docker for reproducible research, with examples from the R environment. ACM SIGOPS Operating Systems Review. 2015;49(1):71–79. doi: 10.1145/2723872.272388. - DOI
-
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43(5):491–498. doi: 10.1038/ng.806. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
