

lncRNA requirements for mouse acute myeloid leukemia and normal differentiation
- PMID: 28875933
- PMCID: PMC5619947
- DOI: 10.7554/eLife.25607
lncRNA requirements for mouse acute myeloid leukemia and normal differentiation
Abstract
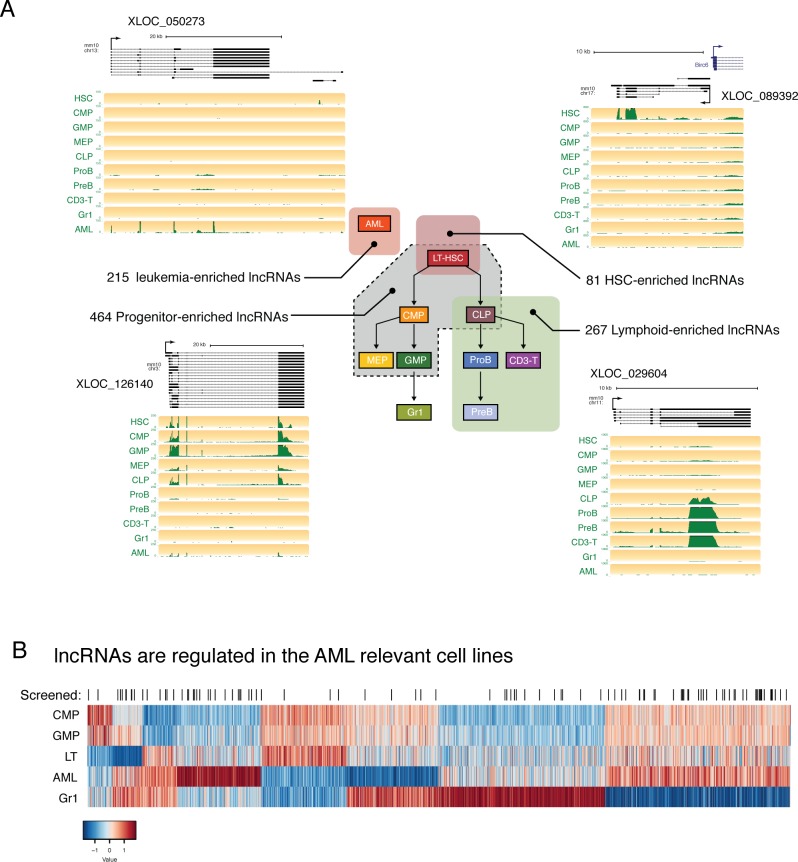
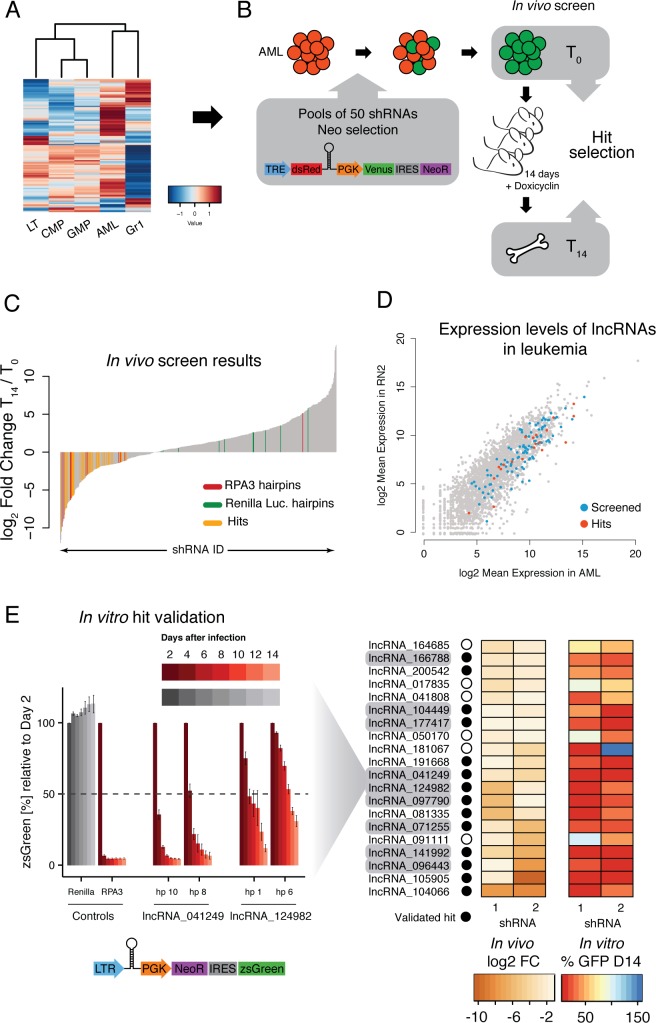
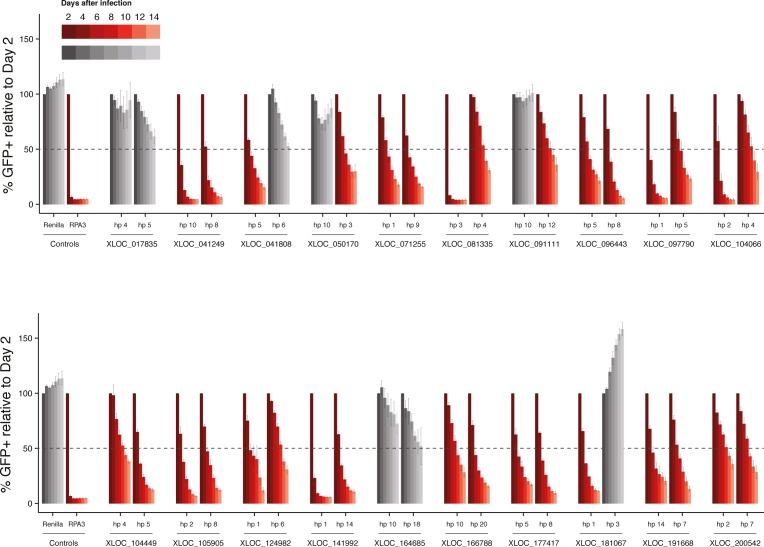
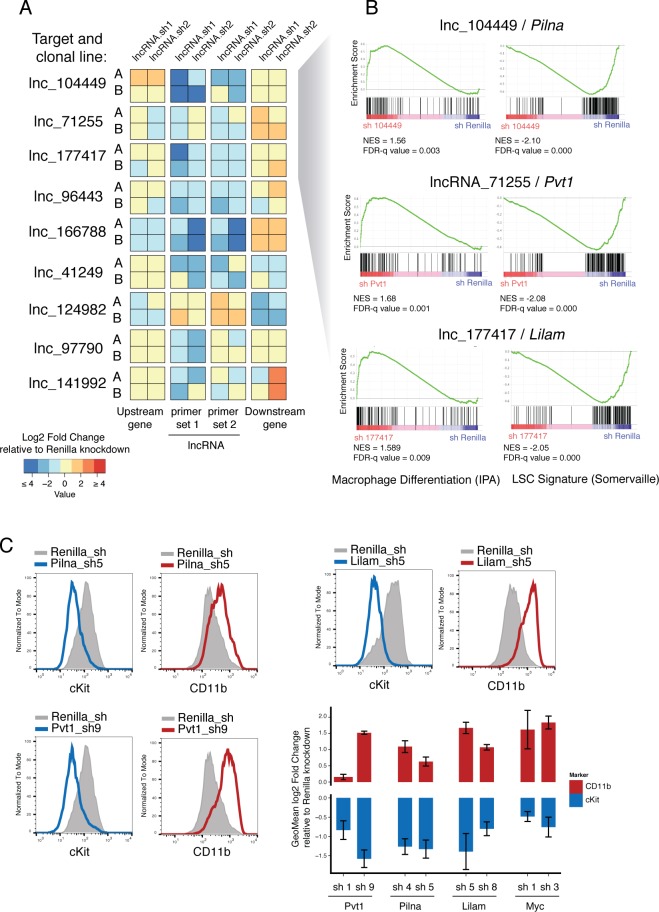
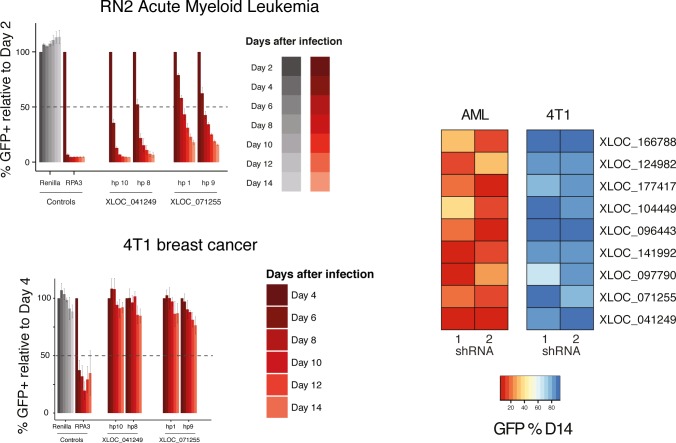
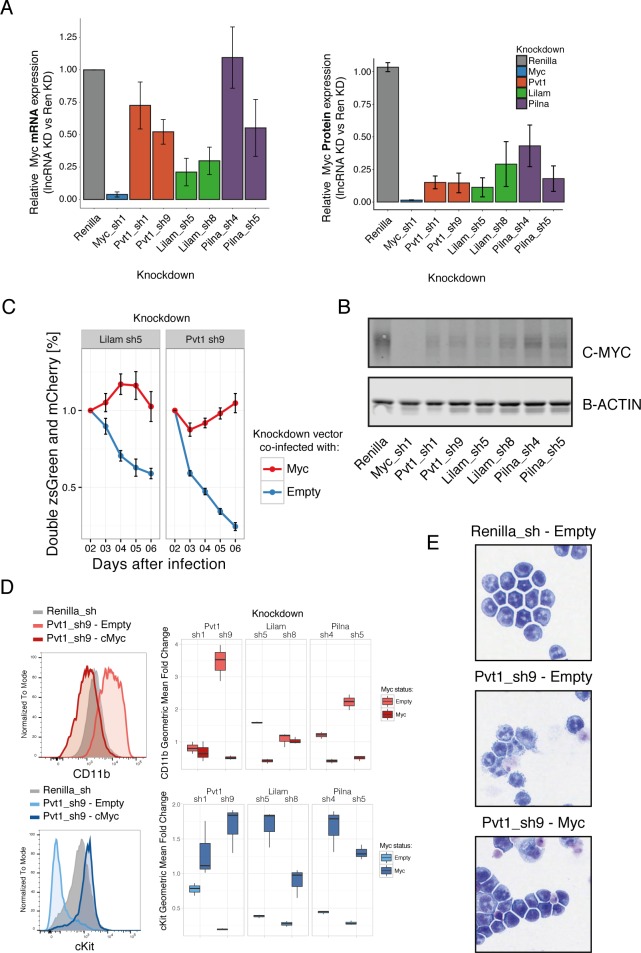
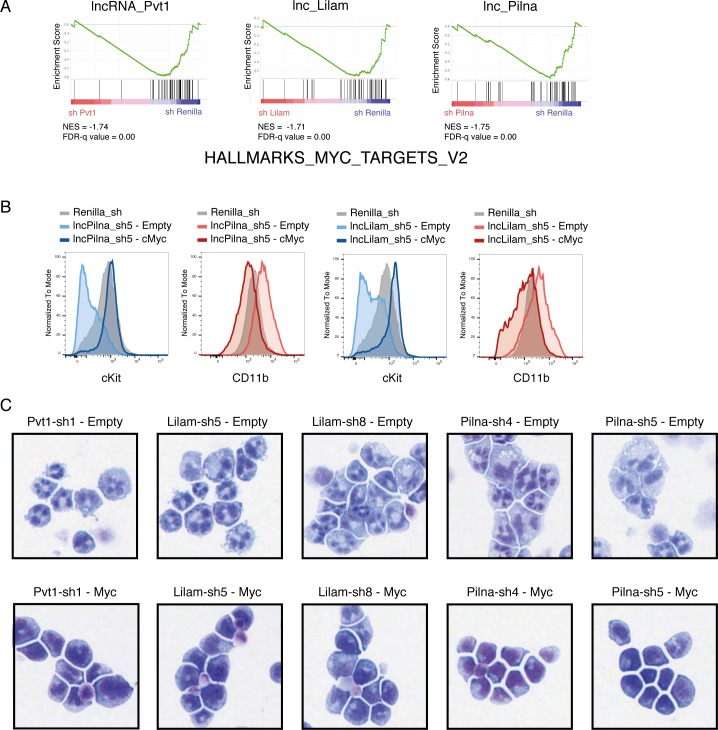
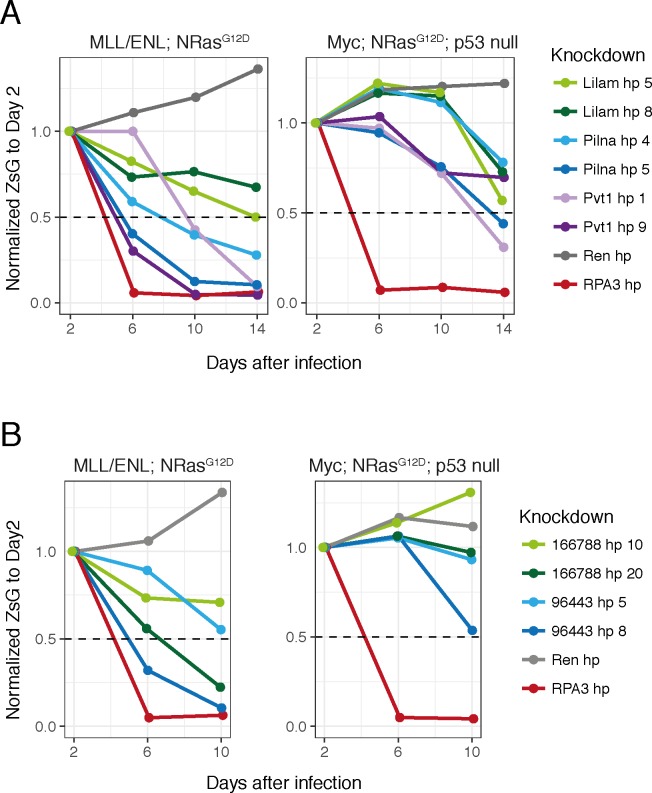
A substantial fraction of the genome is transcribed in a cell-type-specific manner, producing long non-coding RNAs (lncRNAs), rather than protein-coding transcripts. Here, we systematically characterize transcriptional dynamics during hematopoiesis and in hematological malignancies. Our analysis of annotated and de novo assembled lncRNAs showed many are regulated during differentiation and mis-regulated in disease. We assessed lncRNA function via an in vivo RNAi screen in a model of acute myeloid leukemia. This identified several lncRNAs essential for leukemia maintenance, and found that a number act by promoting leukemia stem cell signatures. Leukemia blasts show a myeloid differentiation phenotype when these lncRNAs were depleted, and our data indicates that this effect is mediated via effects on the MYC oncogene. Bone marrow reconstitutions showed that a lncRNA expressed across all progenitors was required for the myeloid lineage, whereas the other leukemia-induced lncRNAs were dispensable in the normal setting.
Keywords: c-Myc; chromosomes; developmental biology; genes; hematopoiesis; leukemia; lncRNA; mouse; stem cells.
Conflict of interest statement
No competing interests declared.
Figures

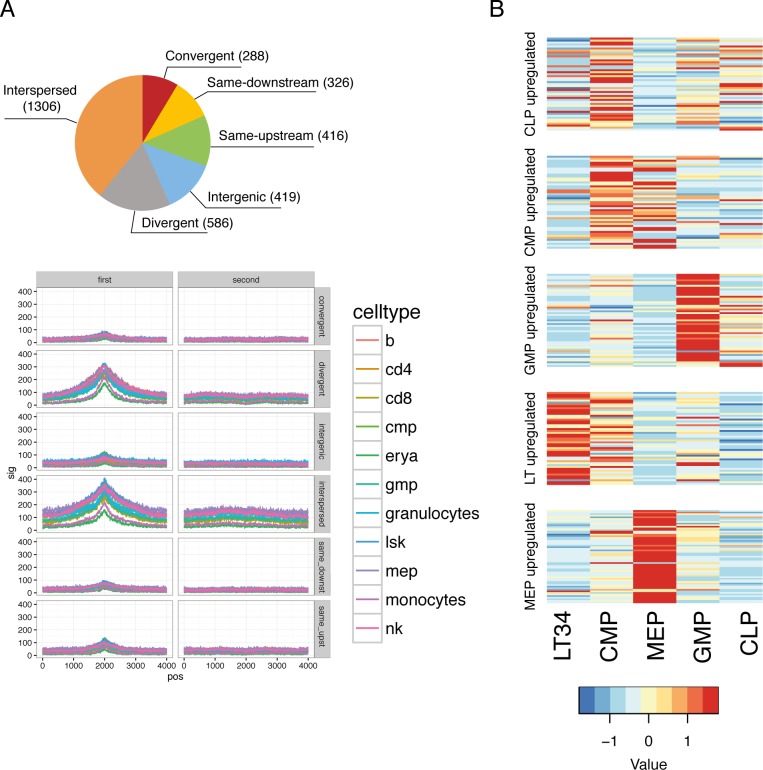
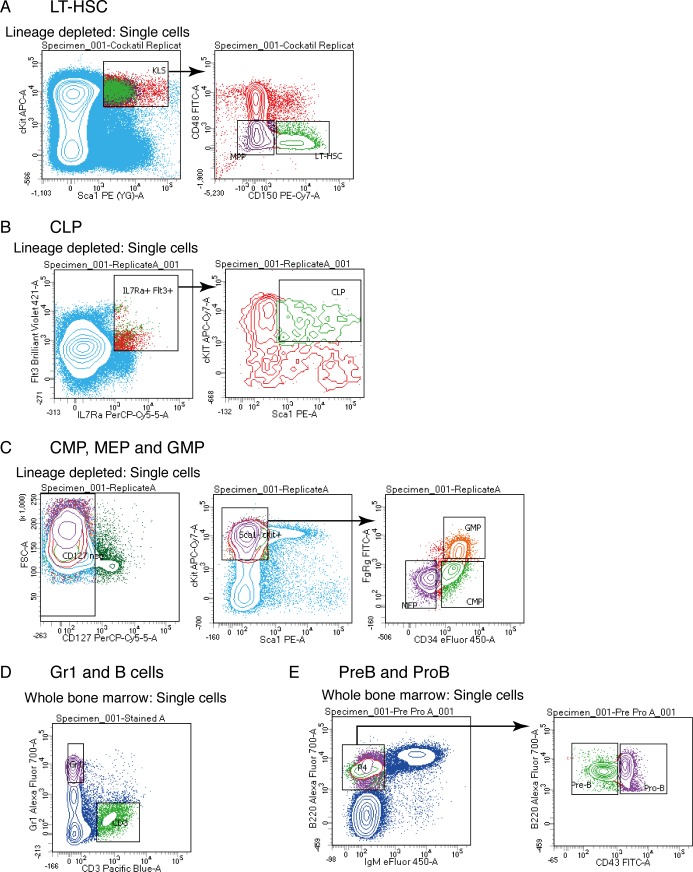
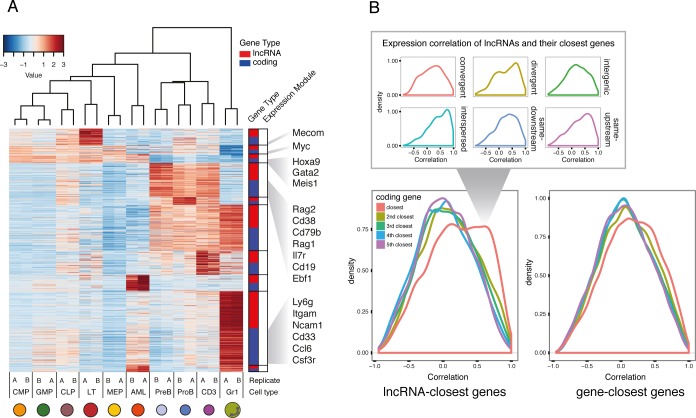
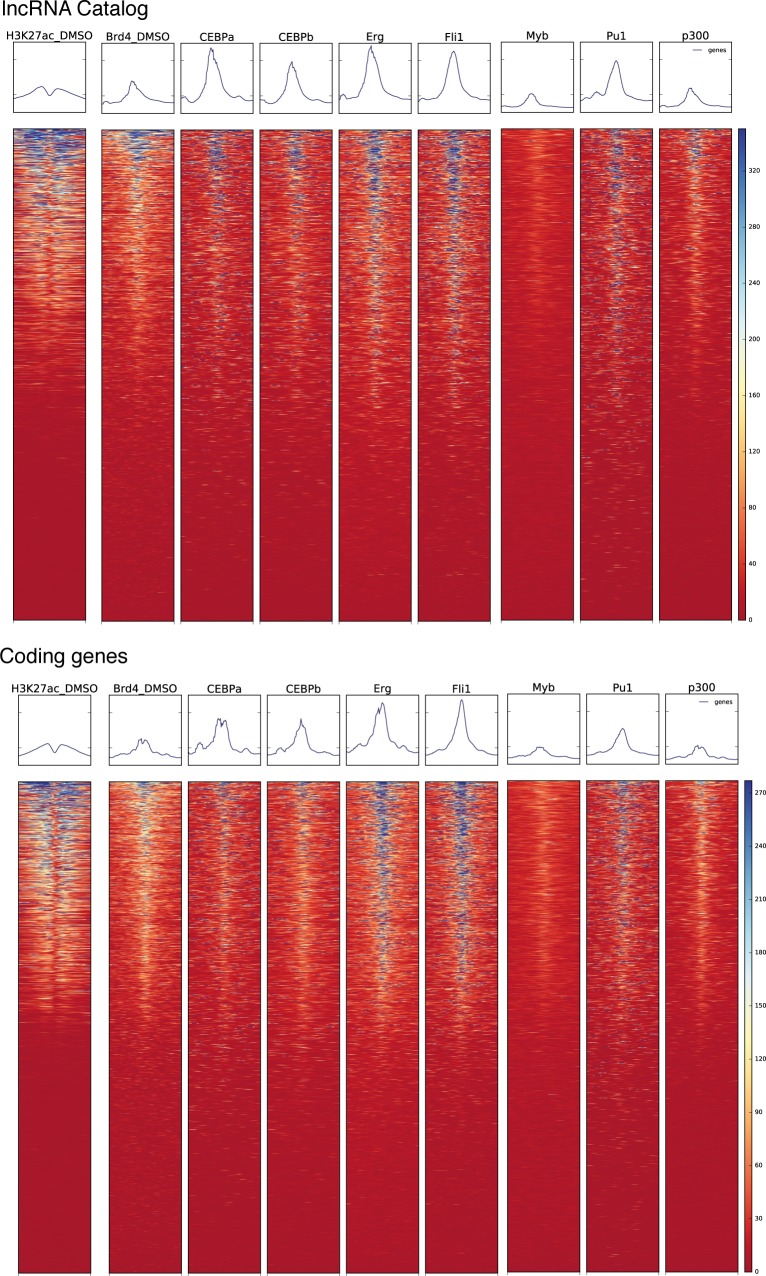
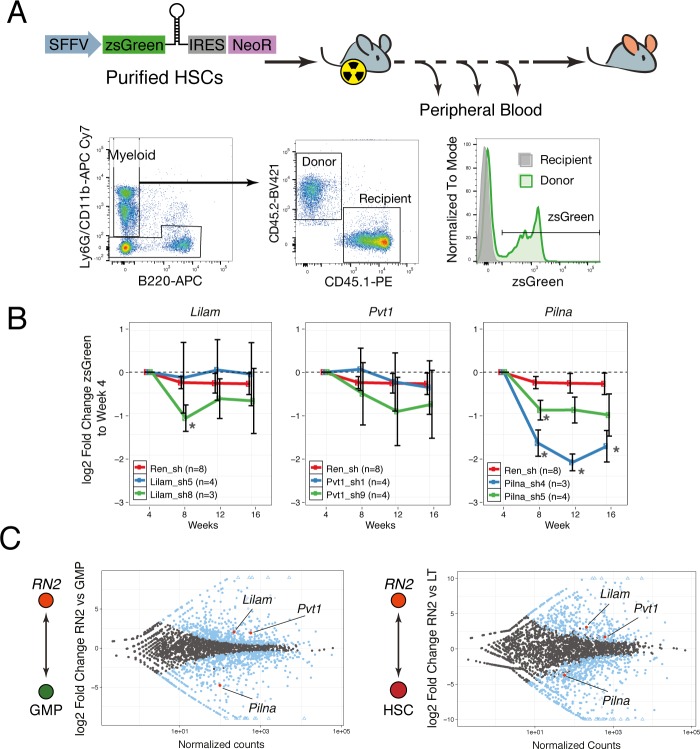
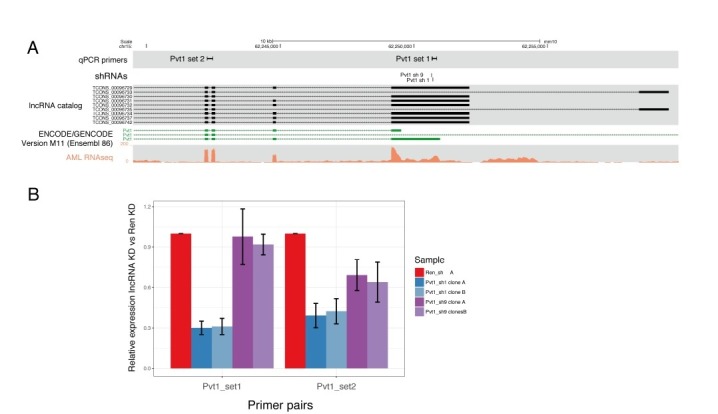
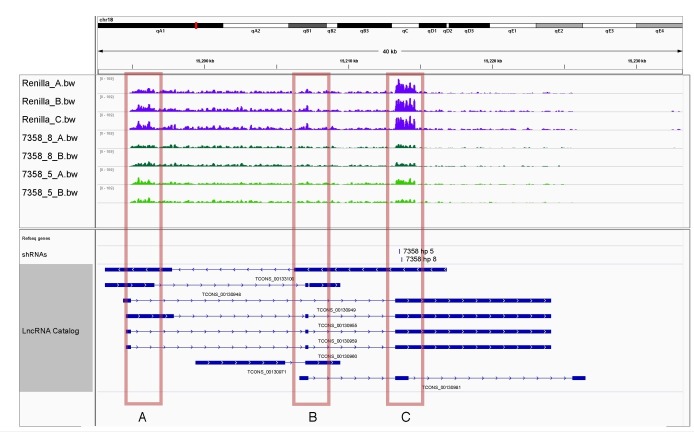
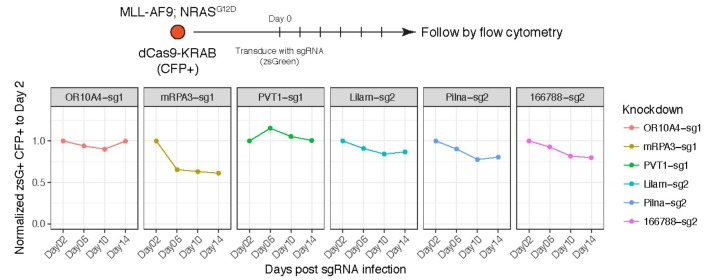
References
-
- Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez-Dominguez JR, Bhatta A, Schattgen SA, McGowan JD, Blin J, Braun JE, Gandhi P, Moore MJ, Chang HY, Lodish HF, Caffrey DR, Fitzgerald KA. A Long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain Inflammation. Cell. 2016;165:1672–1685. doi: 10.1016/j.cell.2016.05.075. - DOI - PMC - PubMed
-
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, Wünsche P, Zeisberger P, Brocks D, Gu L, Herrmann C, Haas S, Essers MA, Brors B, Eils R, Huber W, Milsom MD, Plass C, Krijgsveld J, Trumpp A. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15:507–522. doi: 10.1016/j.stem.2014.07.005. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
