

Acceleration Strategies to Enhance Metabolic Ensemble Modeling Performance
- PMID: 28877496
- PMCID: PMC5611682
- DOI: 10.1016/j.bpj.2017.07.018
Acceleration Strategies to Enhance Metabolic Ensemble Modeling Performance
Abstract
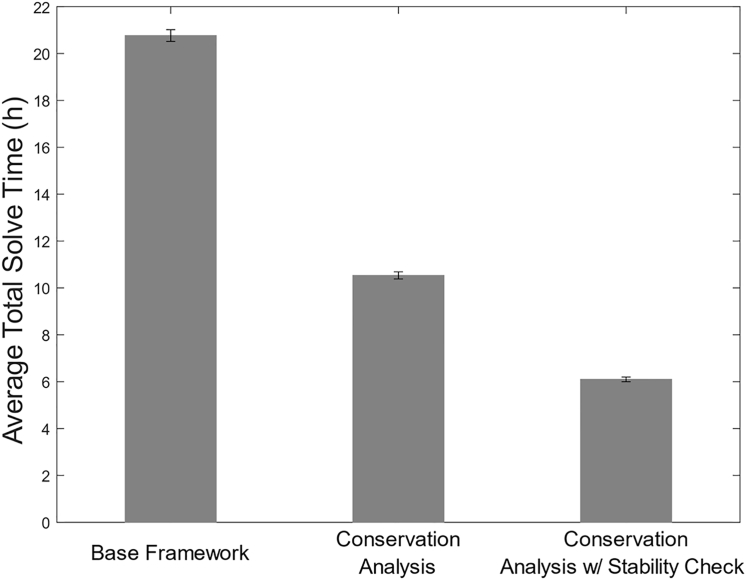
Developing reliable, predictive kinetic models of metabolism is a difficult, yet necessary, priority toward understanding and deliberately altering cellular behavior. Constraint-based modeling has enabled the fields of metabolic engineering and systems biology to make great strides in interrogating cellular metabolism but does not provide sufficient insight into regulation or kinetic limitations of metabolic pathways. Moreover, the growth-optimized assumptions that constraint-based models often rely on do not hold when studying stationary or persistor cell populations. However, developing kinetic models provides many unique challenges, as many of the kinetic parameters and rate laws governing individual enzymes are unknown. Ensemble modeling (EM) was developed to circumnavigate this challenge and effectively sample the large kinetic parameter solution space using consistent experimental datasets. Unfortunately, EM, in its base form, requires long solve times to complete and often leads to unstable kinetic model predictions. Furthermore, these limitations scale prohibitively with increasing model size. As larger metabolic models are developed with increasing genetic information and experimental validation, the demand to incorporate kinetic information increases. Therefore, in this work, we have begun to tackle the challenges of EM by introducing additional steps to the existing method framework specifically through reducing computation time and optimizing parameter sampling. We first reduce the structural complexity of the network by removing dependent species, and second, we sample locally stable parameter sets to reflect realistic biological states of cells. Lastly, we presort the screening data to eliminate the most incorrect predictions in the earliest screening stages, saving further calculations in later stages. Our complementary improvements to this EM framework are easily incorporated into concurrent EM efforts and broaden the application opportunities and accessibility of kinetic modeling across the field.
Copyright © 2017 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures




References
-
- Chowdhury A., Khodayari A., Maranas C.D. Improving prediction fidelity of cellular metabolism with kinetic descriptions. Curr. Opin. Biotechnol. 2015;36:57–64. - PubMed
-
- Srinivasan S., Cluett W.R., Mahadevan R. Constructing kinetic models of metabolism at genome scales: a review. Biotechnol. J. 2015;10:1345–1359. - PubMed
-
- Costa R.S., Hartmann A., Vinga S. Kinetic modeling of cell metabolism for microbial production. J. Biotechnol. 2016;219:126–141. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
