

Enoyl-CoA hydratase-1 regulates mTOR signaling and apoptosis by sensing nutrients
- PMID: 28878358
- PMCID: PMC5587591
- DOI: 10.1038/s41467-017-00489-5
Enoyl-CoA hydratase-1 regulates mTOR signaling and apoptosis by sensing nutrients
Abstract
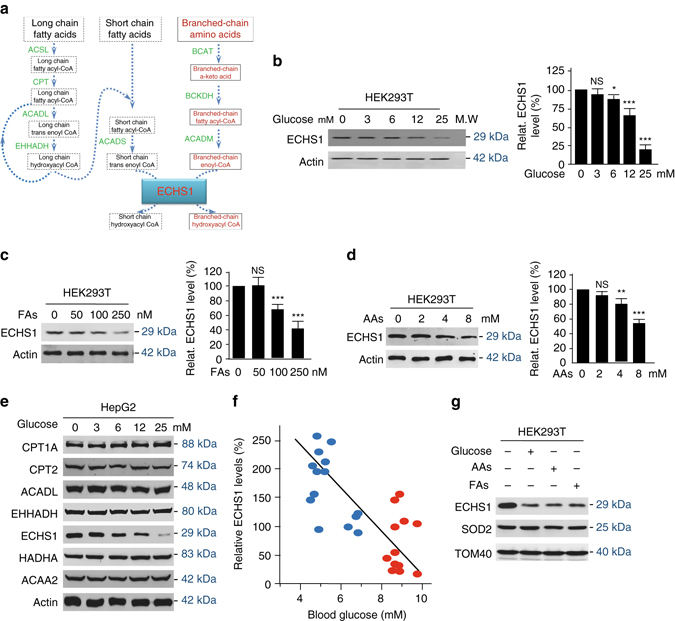
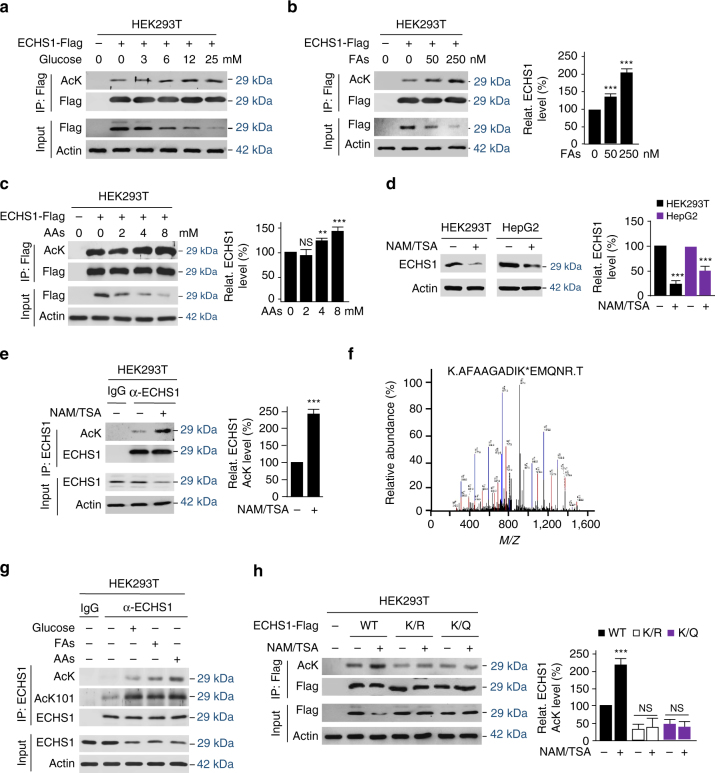
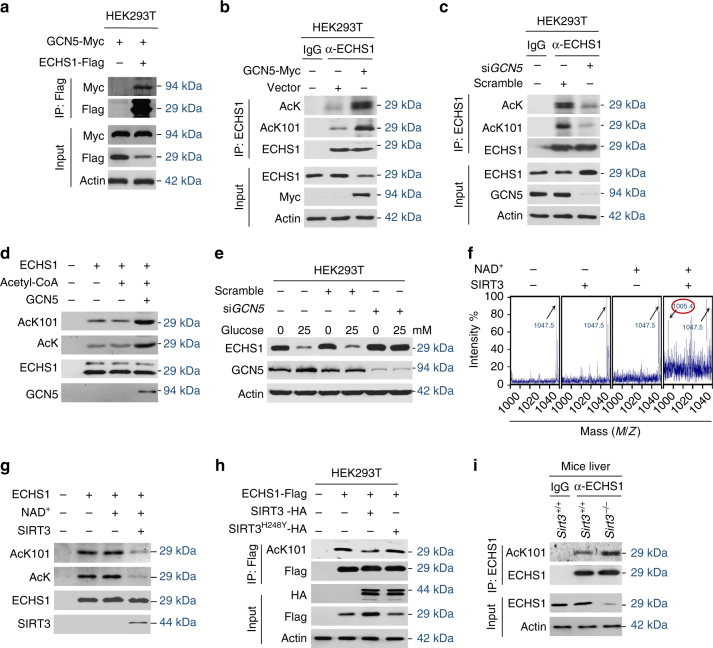
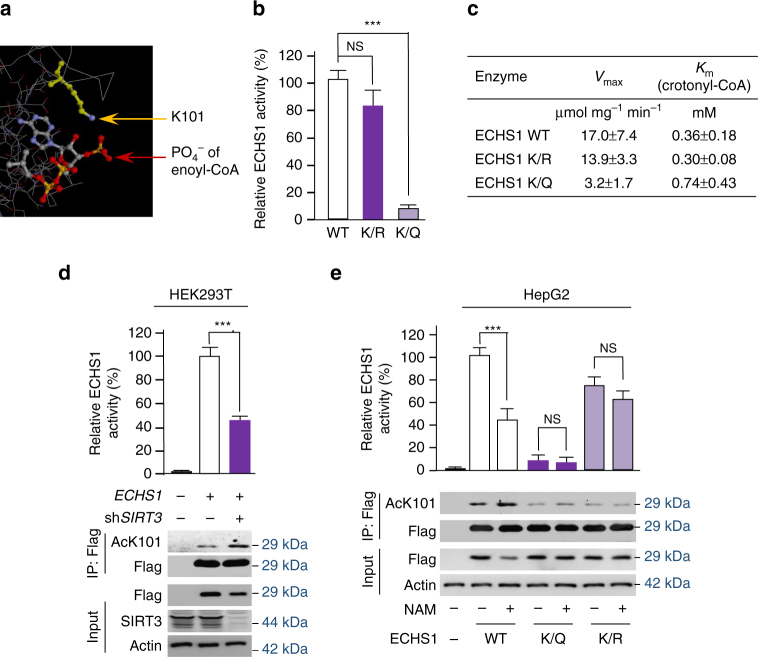
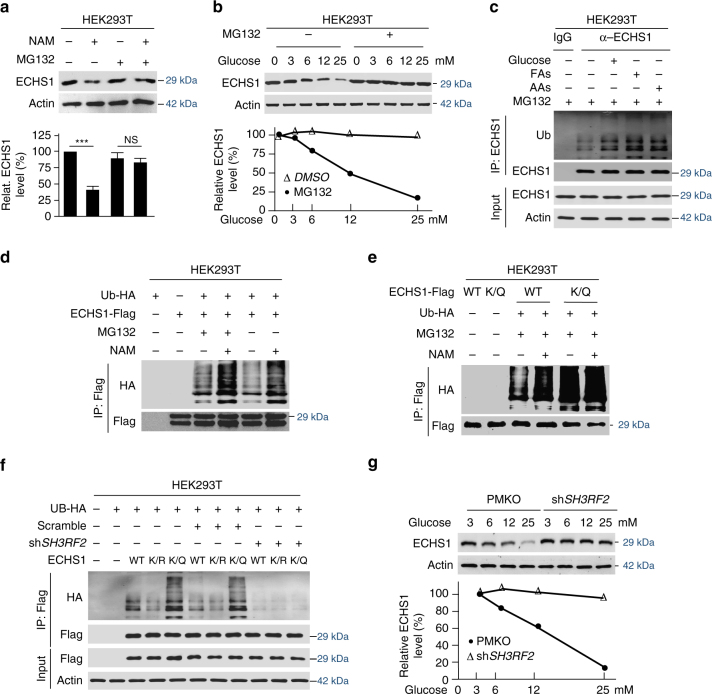
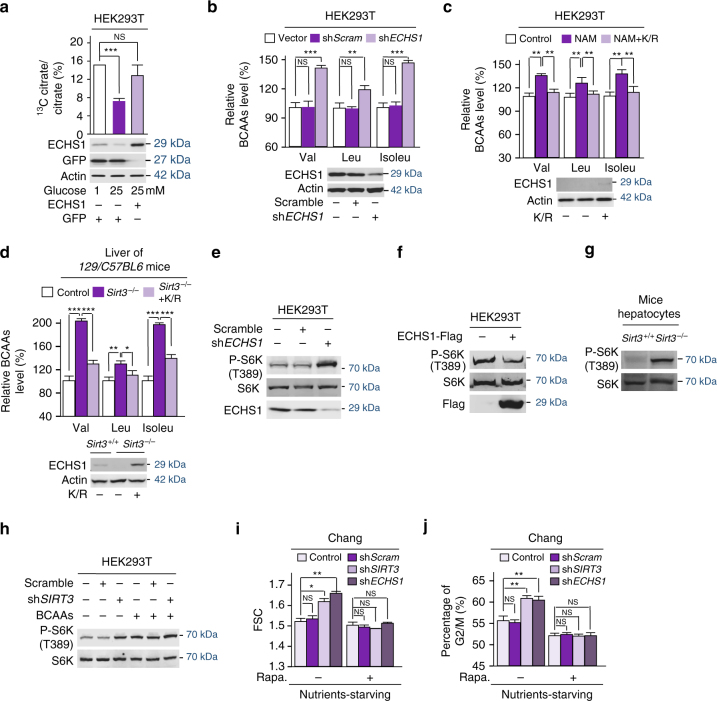
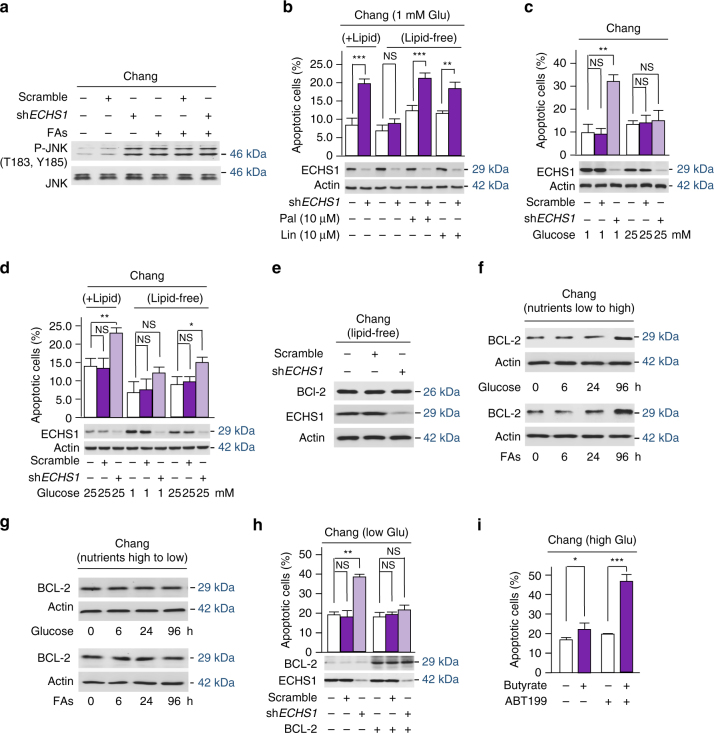
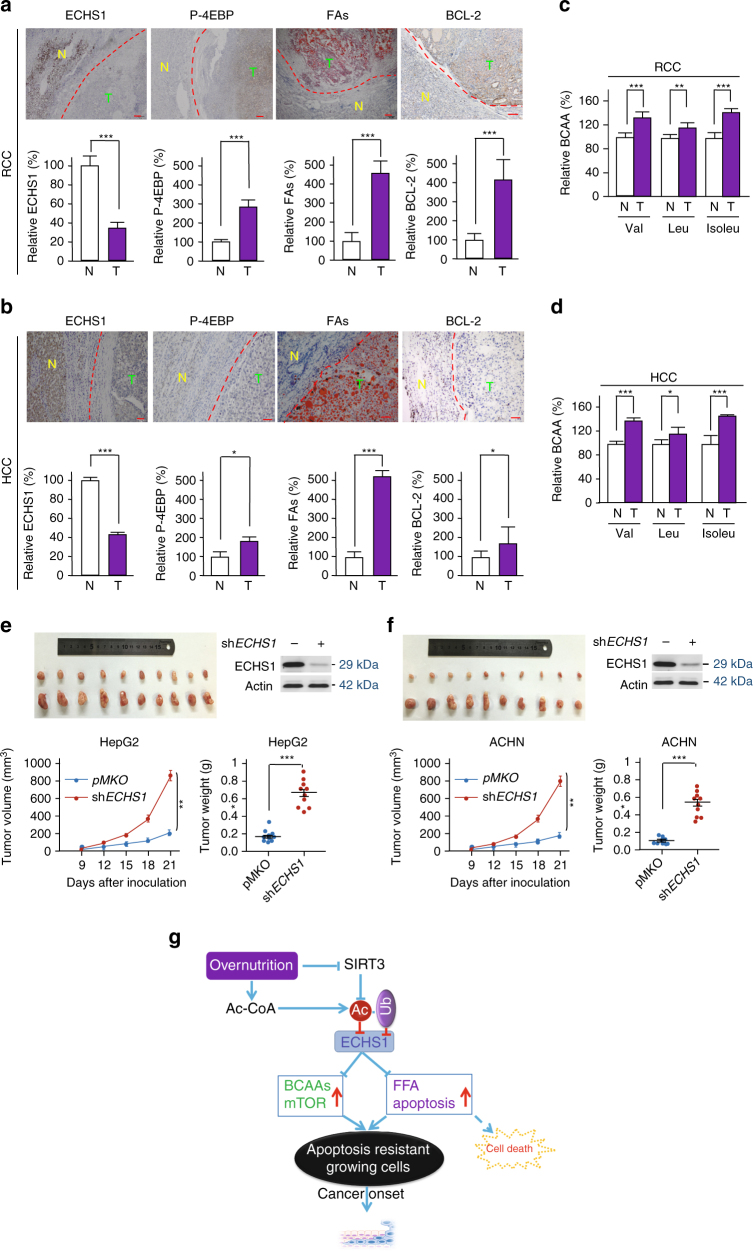
The oncogenic mechanisms of overnutrition, a confirmed independent cancer risk factor, remain poorly understood. Herein, we report that enoyl-CoA hydratase-1 (ECHS1), the enzyme involved in the oxidation of fatty acids (FAs) and branched-chain amino acids (BCAAs), senses nutrients and promotes mTOR activation and apoptotic resistance. Nutrients-promoted acetylation of lys101 of ECHS1 impedes ECHS1 activity by impairing enoyl-CoA binding, promoting ECHS1 degradation and blocking its mitochondrial translocation through inducing ubiquitination. As a result, nutrients induce the accumulation of BCAAs and FAs that activate mTOR signaling and stimulate apoptosis, respectively. The latter was overcome by selection of BCL-2 overexpressing cells under overnutrition conditions. The oncogenic effects of nutrients were reversed by SIRT3, which deacetylates lys101 acetylation. Severely decreased ECHS1, accumulation of BCAAs and FAs, activation of mTOR and overexpression of BCL-2 were observed in cancer tissues from metabolic organs. Our results identified ECHS1, a nutrients-sensing protein that transforms nutrient signals into oncogenic signals.Overnutrition has been linked to increased risk of cancer. Here, the authors show that exceeding nutrients suppress Enoyl-CoA hydratase-1 (ECHS1) activity by inducing its acetylation resulting in accumulation of fatty acids and branched-chain amino acids and oncogenic mTOR activation.
Conflict of interest statement
The authors declare no competing financial interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
