

Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand
- PMID: 28878620
- PMCID: PMC5572252
- DOI: 10.3389/fnmol.2017.00263
Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand
Abstract
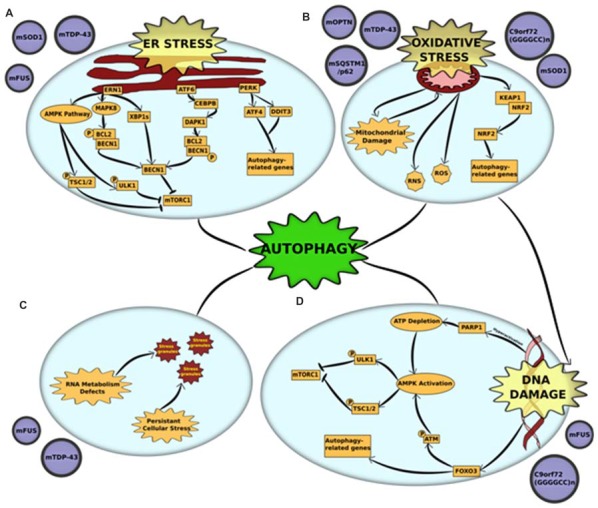
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder that results from the loss of upper and lower motor neurons. One of the key pathological hallmarks in diseased neurons is the mislocalization of disease-associated proteins and the formation of cytoplasmic aggregates of these proteins and their interactors due to defective protein quality control. This apparent imbalance in the cellular protein homeostasis could be a crucial factor in causing motor neuron death in the later stages of the disease in patients. Autophagy is a major protein degradation pathway that is involved in the clearance of protein aggregates and damaged organelles. Abnormalities in autophagy have been observed in numerous neurodegenerative disorders, including ALS. In this review, we discuss the contribution of autophagy dysfunction in various in vitro and in vivo models of ALS. Furthermore, we examine the crosstalk between autophagy and other cellular stresses implicated in ALS pathogenesis and the therapeutic implications of regulating autophagy in ALS.
Keywords: ALS; C9orf72; FUS; SOD1; TDP-43; autophagy; motor neuron disease; neurodegeneration.
Figures
References
-
- Abe K., Itoyama Y., Sobue G., Tsuji S., Aoki M., Doyu M., et al. . (2014). Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 610–617. 10.3109/21678421.2014.959024 - DOI - PMC - PubMed
-
- Al-Sarraj S., King A., Troakes C., Smith B., Maekawa S., Bodi I., et al. . (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. 10.1007/s00401-011-0911-2 - DOI - PubMed
-
- Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., et al. . (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. 10.1016/j.bbrc.2006.10.093 - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous