

Orthologous Matrix (OMA) algorithm 2.0: more robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference
- PMID: 28881964
- PMCID: PMC5870696
- DOI: 10.1093/bioinformatics/btx229
Orthologous Matrix (OMA) algorithm 2.0: more robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference
Abstract
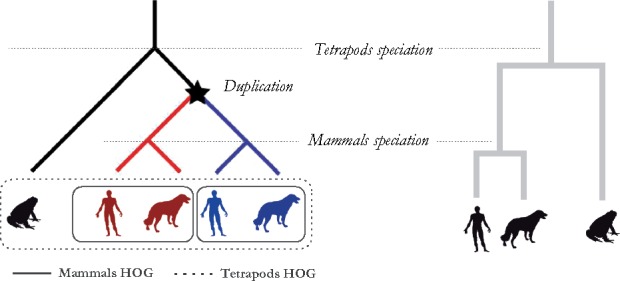
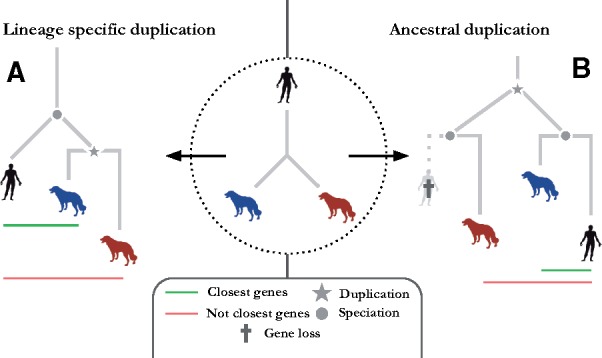
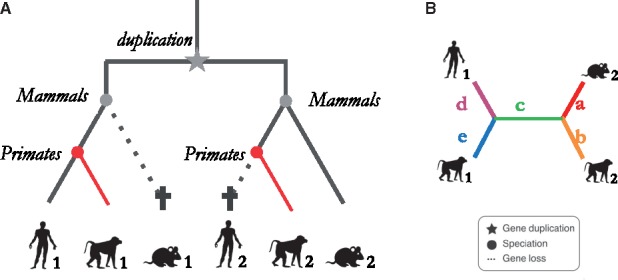
Motivation: Accurate orthology inference is a fundamental step in many phylogenetics and comparative analysis. Many methods have been proposed, including OMA (Orthologous MAtrix). Yet substantial challenges remain, in particular in coping with fragmented genes or genes evolving at different rates after duplication, and in scaling to large datasets. With more and more genomes available, it is necessary to improve the scalability and robustness of orthology inference methods.
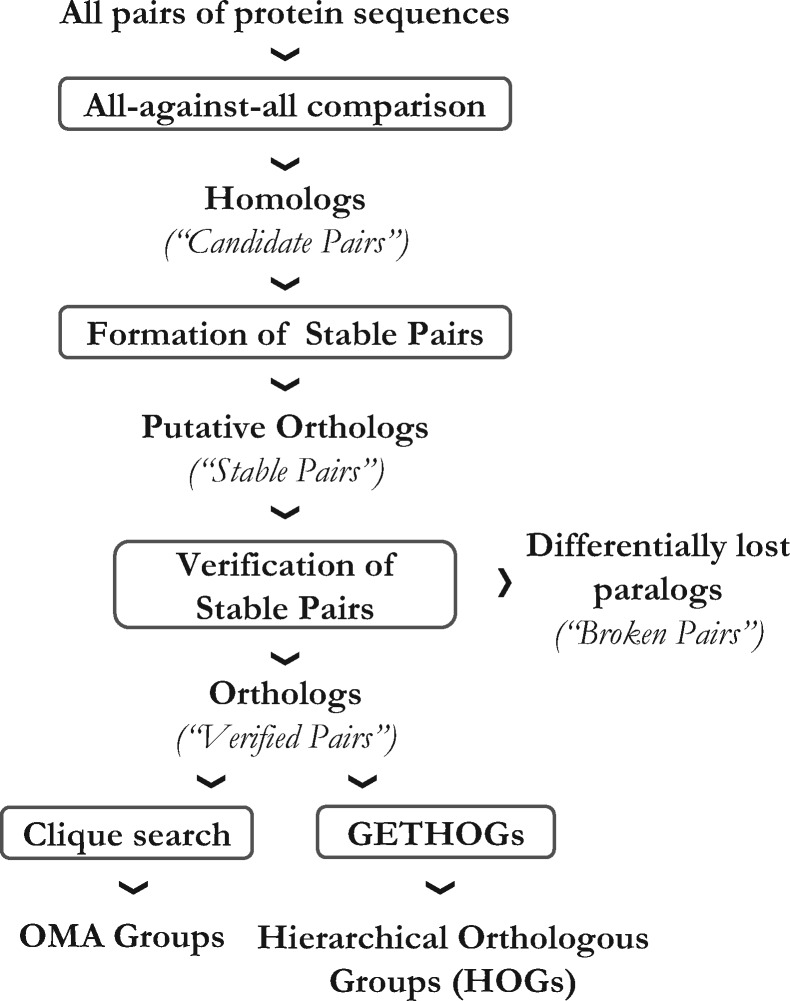
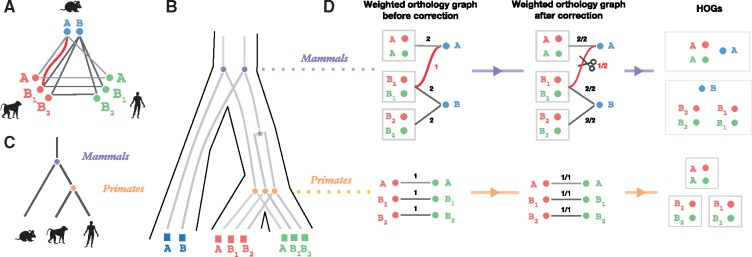
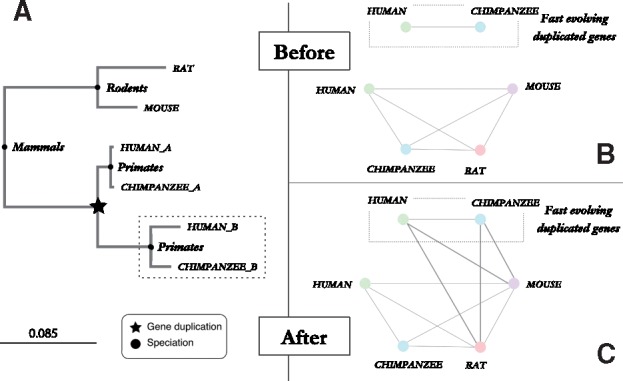
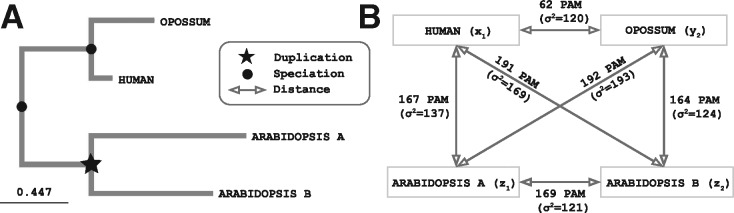
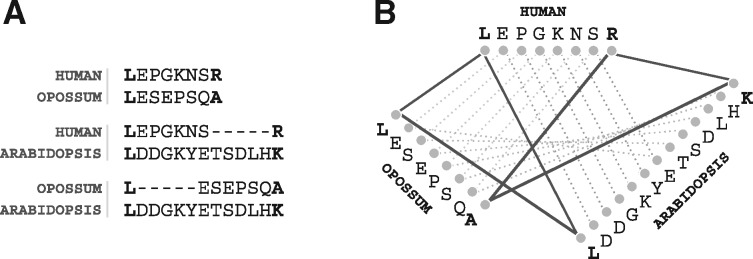
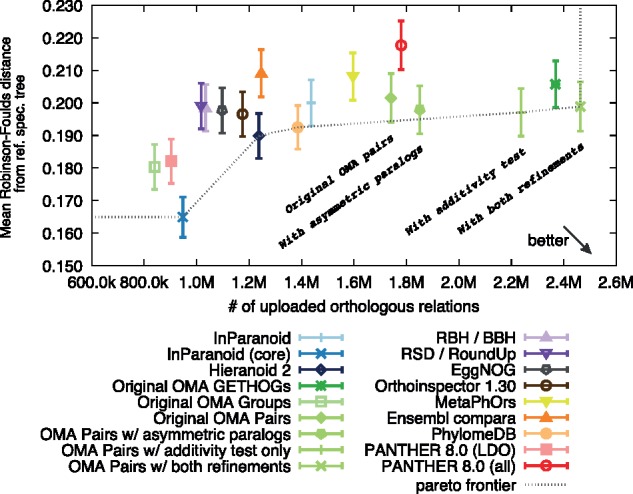
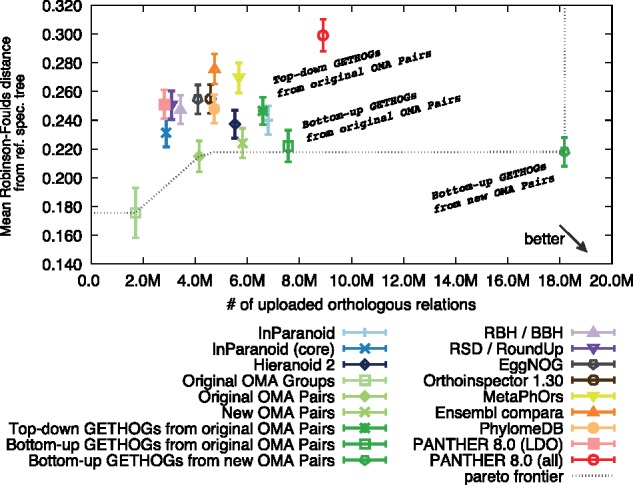
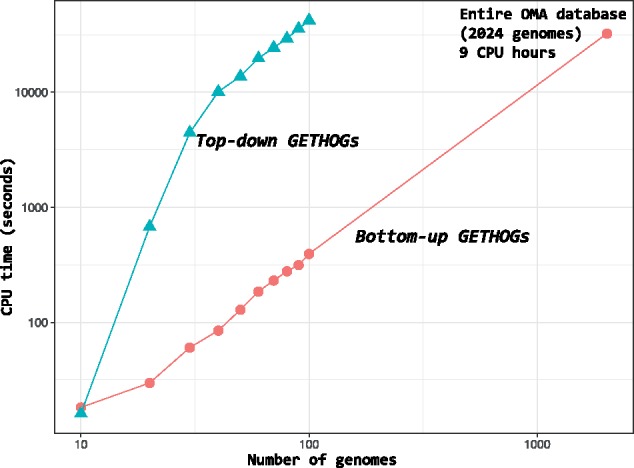
Results: We present improvements in the OMA algorithm: (i) refining the pairwise orthology inference step to account for same-species paralogs evolving at different rates, and (ii) minimizing errors in the pairwise orthology verification step by testing the consistency of pairwise distance estimates, which can be problematic in the presence of fragmentary sequences. In addition we introduce a more scalable procedure for hierarchical orthologous group (HOG) clustering, which are several orders of magnitude faster on large datasets. Using the Quest for Orthologs consortium orthology benchmark service, we show that these changes translate into substantial improvement on multiple empirical datasets.
Availability and implementation: This new OMA 2.0 algorithm is used in the OMA database ( http://omabrowser.org ) from the March 2017 release onwards, and can be run on custom genomes using OMA standalone version 2.0 and above ( http://omabrowser.org/standalone ).
Contact: christophe.dessimoz@unil.ch or adrian.altenhoff@inf.ethz.ch.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com
Figures

References
-
- Ballesteros J.A., Hormiga G. (2016) A new orthology assessment method for phylogenomic data: unrooted phylogenetic orthology. Mol. Biol. Evol., 33, 2481. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
