

Molecular signatures that can be transferred across different omics platforms
- PMID: 28881975
- PMCID: PMC5870545
- DOI: 10.1093/bioinformatics/btx241
Molecular signatures that can be transferred across different omics platforms
Erratum in
-
Molecular signatures that can be transferred across different omics platforms.Bioinformatics. 2017 Sep 1;33(17):2790. doi: 10.1093/bioinformatics/btx488. Bioinformatics. 2017. PMID: 28903540 Free PMC article. No abstract available.
Abstract
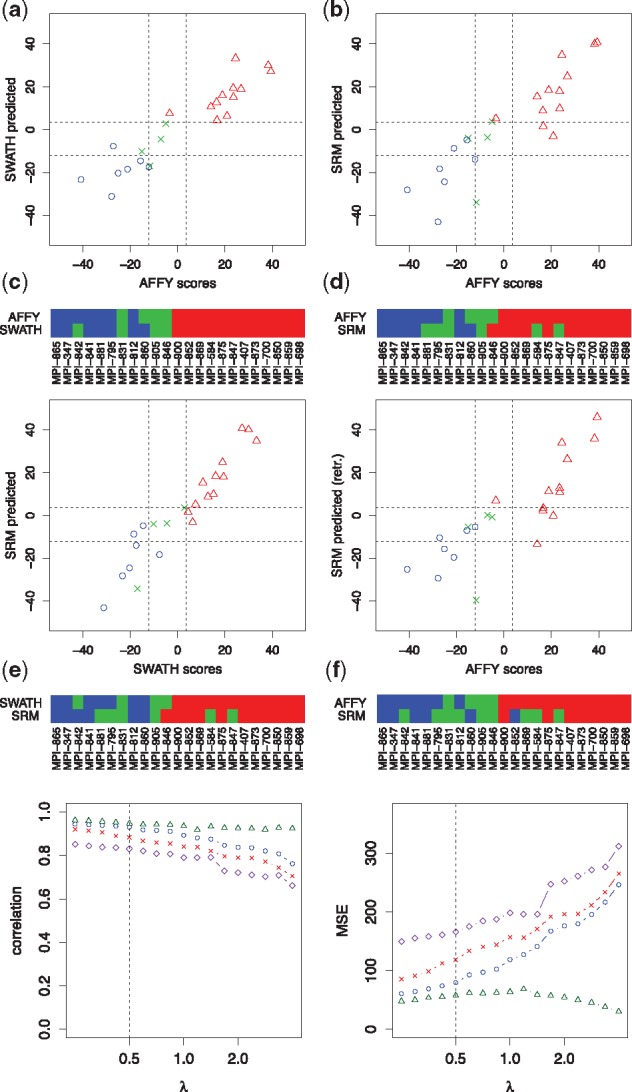
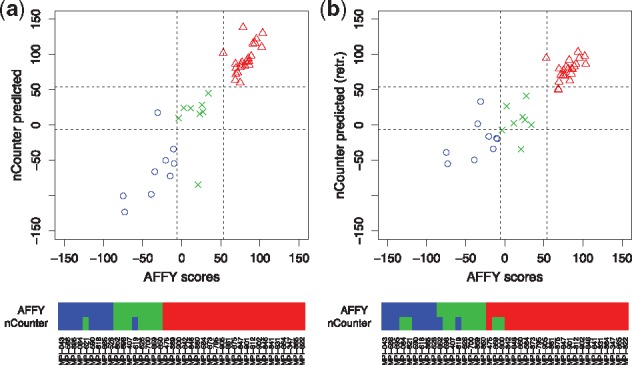
Motivation: Molecular signatures for treatment recommendations are well researched. Still it is challenging to apply them to data generated by different protocols or technical platforms.
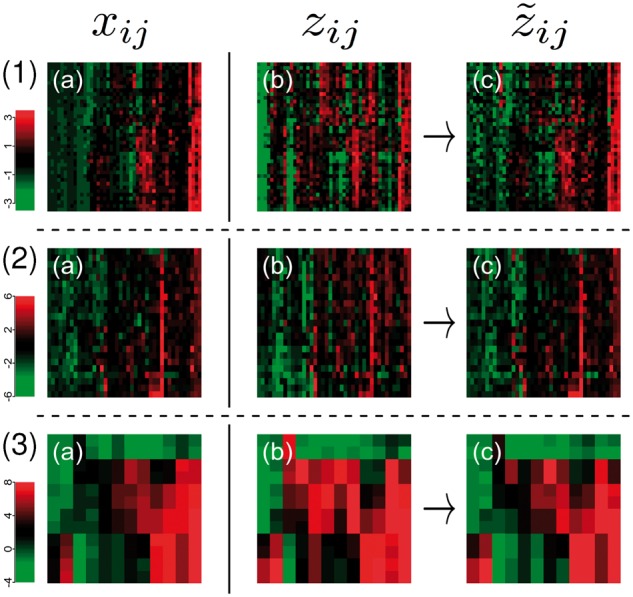
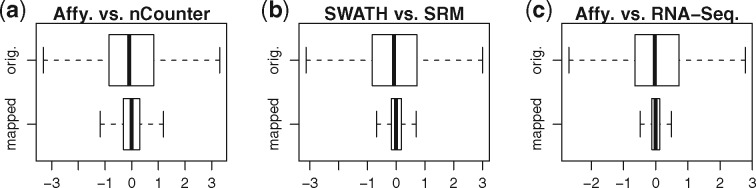
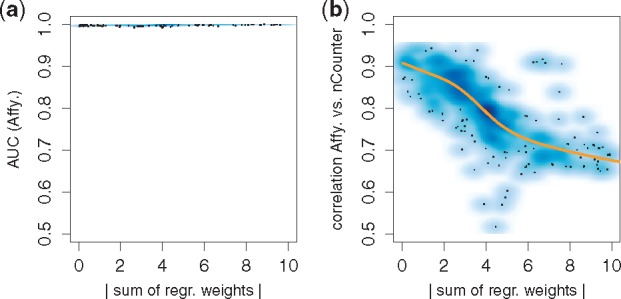
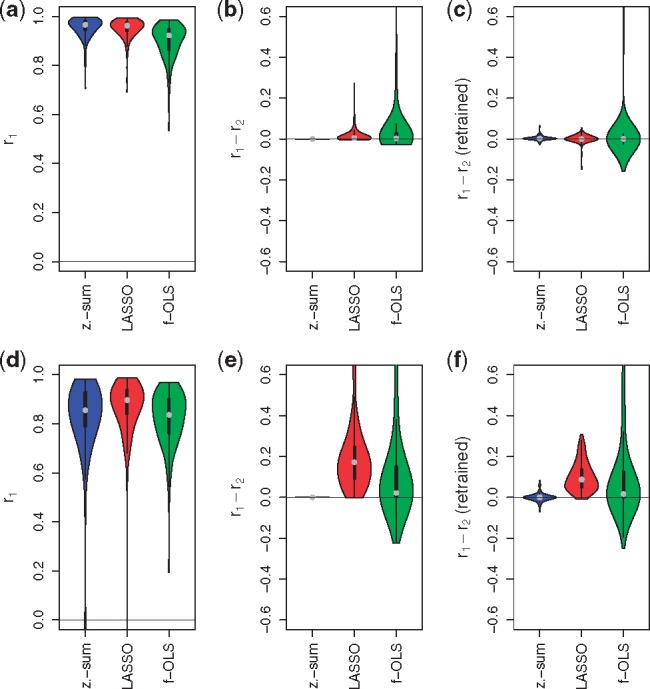
Results: We analyzed paired data for the same tumors (Burkitt lymphoma, diffuse large B-cell lymphoma) and features that had been generated by different experimental protocols and analytical platforms including the nanoString nCounter and Affymetrix Gene Chip transcriptomics as well as the SWATH and SRM proteomics platforms. A statistical model that assumes independent sample and feature effects accounted for 69-94% of technical variability. We analyzed how variability is propagated through linear signatures possibly affecting predictions and treatment recommendations. Linear signatures with feature weights adding to zero were substantially more robust than unbalanced signatures. They yielded consistent predictions across data from different platforms, both for transcriptomics and proteomics data. Similarly stable were their predictions across data from fresh frozen and matching formalin-fixed paraffin-embedded human tumor tissue.
Availability and implementation: The R-package 'zeroSum' can be downloaded at https://github.com/rehbergT/zeroSum . Complete data and R codes necessary to reproduce all our results can be received from the authors upon request.
Contact: rainer.spang@ur.de.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com
Figures
References
-
- Alizadeh A.A. et al. (2000) Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature, 403, 503–511. - PubMed
-
- Altenbuchinger M. et al. (2017) Reference point insensitive molecular data analysis. Bioinformatics, 33, 219–226. - PubMed
-
- Dave S.S. et al. (2006) Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med., 354, 2431–2442. - PubMed
-
- Faktor J. et al. (2016) Comparison of targeted proteomics approaches for detecting and quantifying proteins derived from human cancer tissues. Proteomics, 17, S.1600323. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
