

Tau exacerbates excitotoxic brain damage in an animal model of stroke
- PMID: 28883427
- PMCID: PMC5589746
- DOI: 10.1038/s41467-017-00618-0
Tau exacerbates excitotoxic brain damage in an animal model of stroke
Abstract
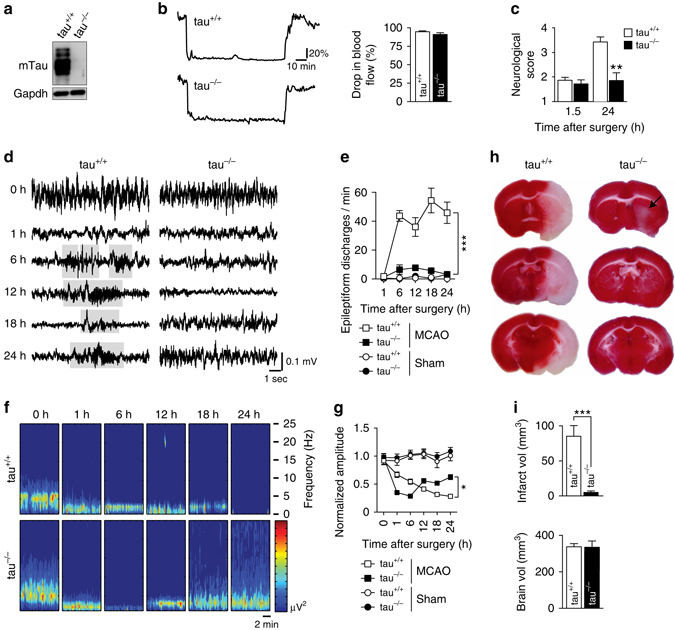
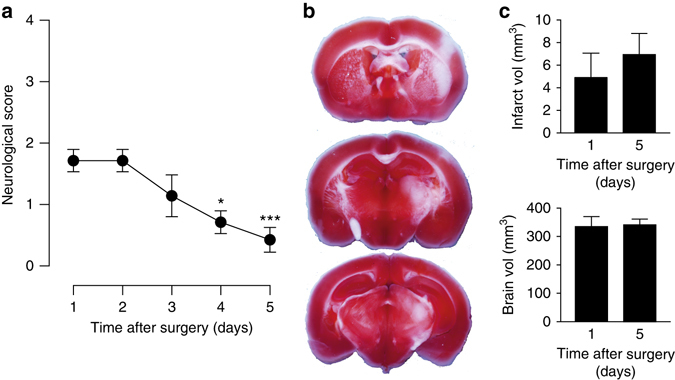
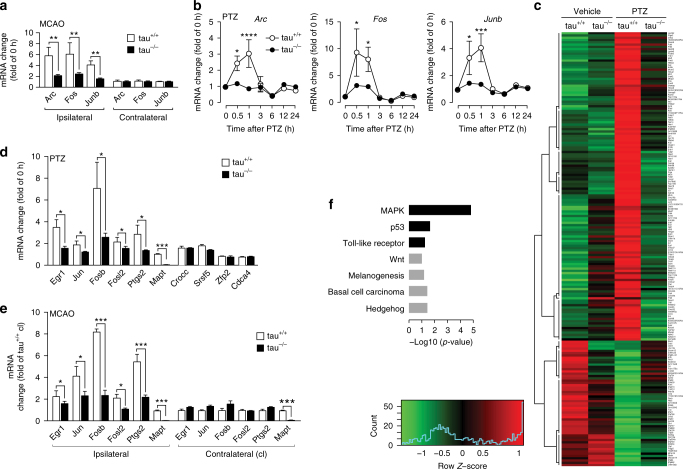
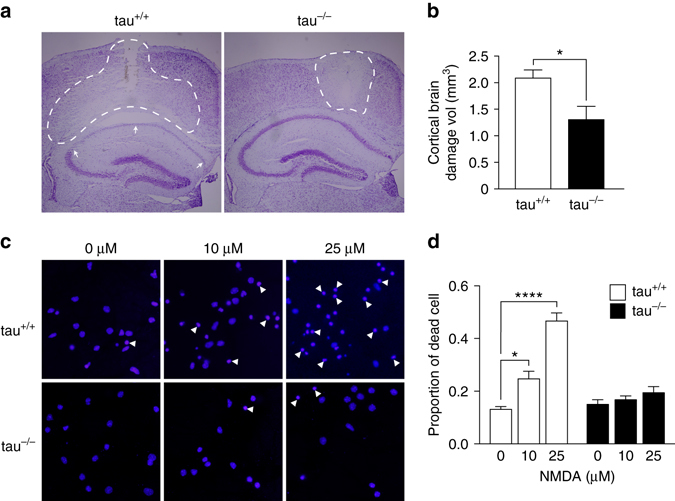
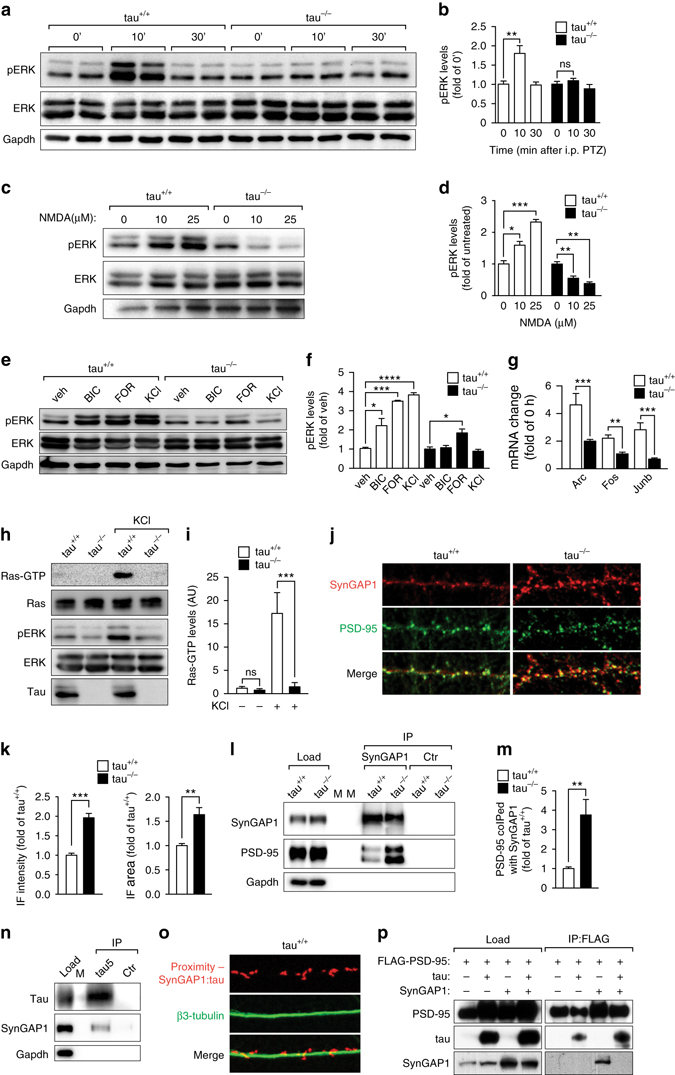
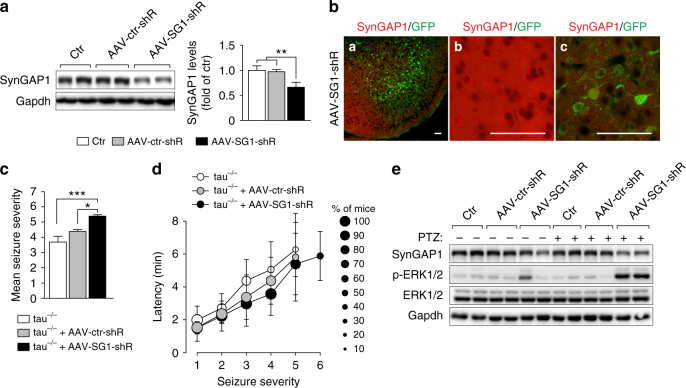
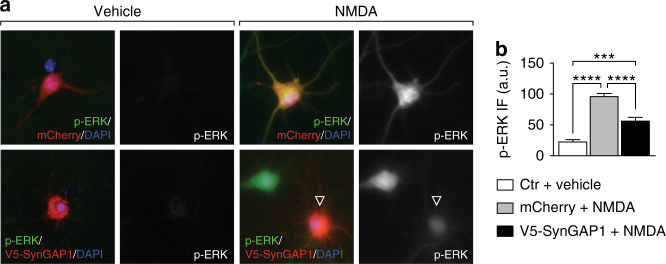
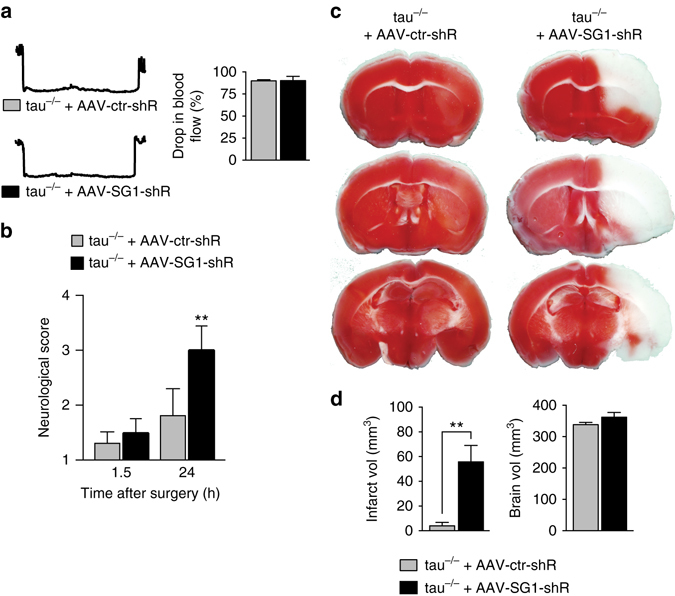
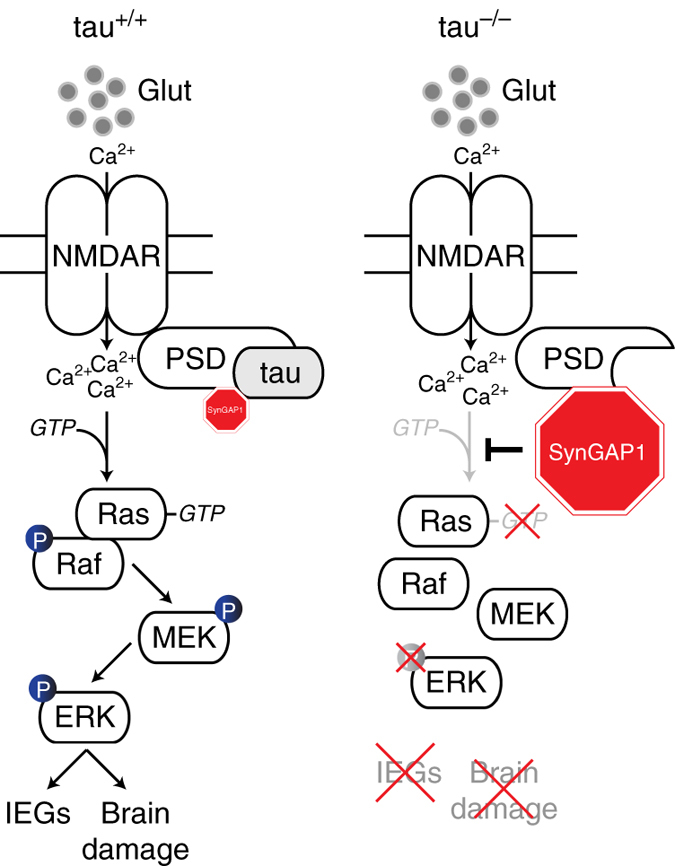
Neuronal excitotoxicity induced by aberrant excitation of glutamatergic receptors contributes to brain damage in stroke. Here we show that tau-deficient (tau-/-) mice are profoundly protected from excitotoxic brain damage and neurological deficits following experimental stroke, using a middle cerebral artery occlusion with reperfusion model. Mechanistically, we show that this protection is due to site-specific inhibition of glutamate-induced and Ras/ERK-mediated toxicity by accumulation of Ras-inhibiting SynGAP1, which resides in a post-synaptic complex with tau. Accordingly, reducing SynGAP1 levels in tau-/- mice abolished the protection from pharmacologically induced excitotoxicity and middle cerebral artery occlusion-induced brain damage. Conversely, over-expression of SynGAP1 prevented excitotoxic ERK activation in wild-type neurons. Our findings suggest that tau mediates excitotoxic Ras/ERK signaling by controlling post-synaptic compartmentalization of SynGAP1.Excitotoxicity contributes to neuronal injury following stroke. Here the authors show that tau promotes excitotoxicity by a post-synaptic mechanism, involving site-specific control of ERK activation, in a mouse model of stroke.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

Comment in
-
Stroke: Tau - a new target in acute brain ischaemia.Nat Rev Neurol. 2017 Nov;13(11):639. doi: 10.1038/nrneurol.2017.141. Epub 2017 Sep 29. Nat Rev Neurol. 2017. PMID: 28960188 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
