

Alterations of the Gut Microbiome in Hypertension
- PMID: 28884091
- PMCID: PMC5573791
- DOI: 10.3389/fcimb.2017.00381
Alterations of the Gut Microbiome in Hypertension
Abstract
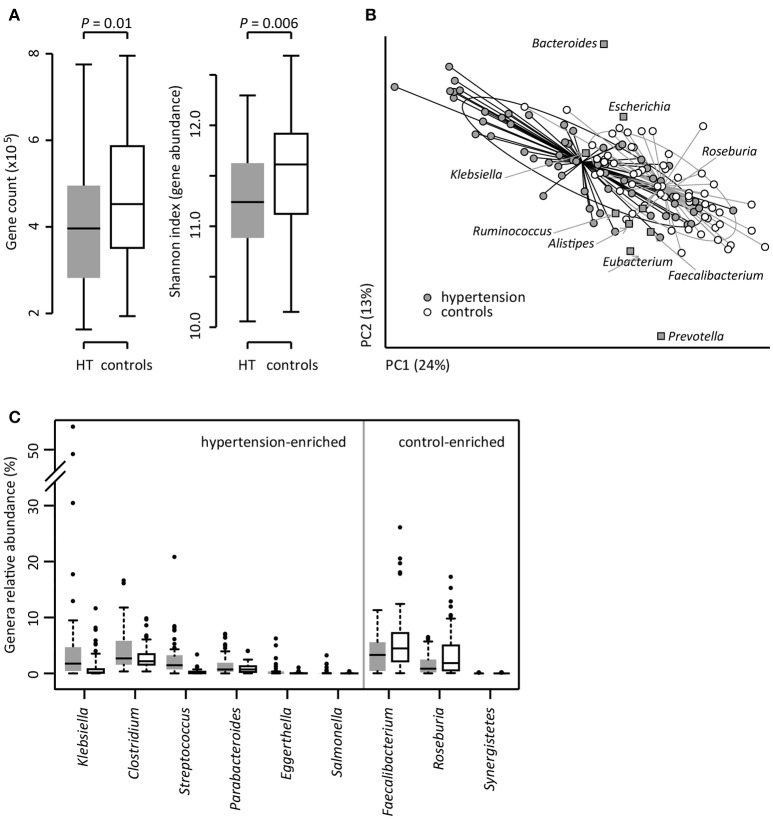
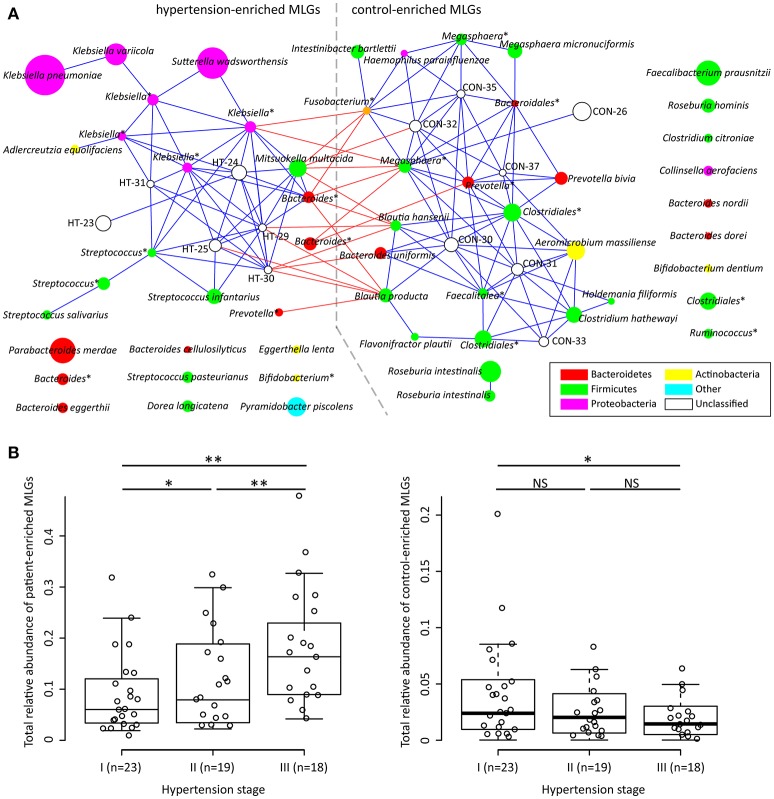
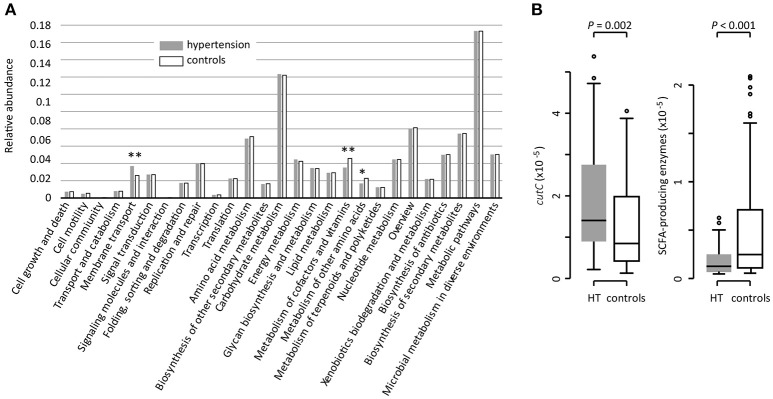
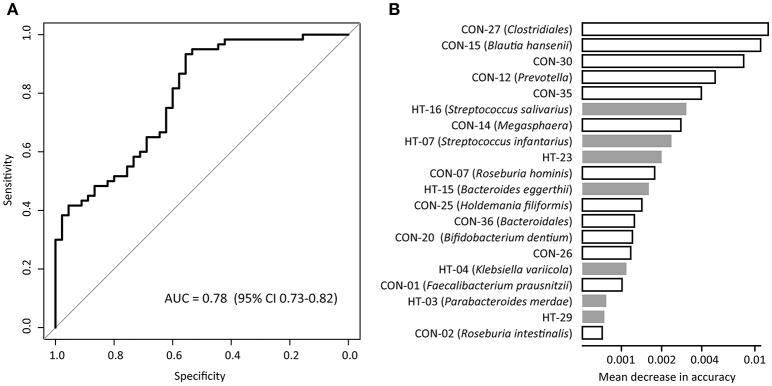
Introduction: Human gut microbiota is believed to be directly or indirectly involved in cardiovascular diseases and hypertension. However, the identification and functional status of the hypertension-related gut microbe(s) have not yet been surveyed in a comprehensive manner. Methods: Here we characterized the gut microbiome in hypertension status by comparing fecal samples of 60 patients with primary hypertension and 60 gender-, age-, and body weight-matched healthy controls based on whole-metagenome shotgun sequencing. Results: Hypertension implicated a remarkable gut dysbiosis with significant reduction in within-sample diversity and shift in microbial composition. Metagenome-wide association study (MGWAS) revealed 53,953 microbial genes that differ in distribution between the patients and healthy controls (false discovery rate, 0.05) and can be grouped into 68 clusters representing bacterial species. Opportunistic pathogenic taxa, such as, Klebsiella spp., Streptococcus spp., and Parabacteroides merdae were frequently distributed in hypertensive gut microbiome, whereas the short-chain fatty acid producer, such as, Roseburia spp. and Faecalibacterium prausnitzii, were higher in controls. The number of hypertension-associated species also showed stronger correlation to the severity of disease. Functionally, the hypertensive gut microbiome exhibited higher membrane transport, lipopolysaccharide biosynthesis and steroid degradation, while in controls the metabolism of amino acid, cofactors and vitamins was found to be higher. We further provided the microbial markers for disease discrimination and achieved an area under the receiver operator characteristic curve (AUC) of 0.78, demonstrating the potential of gut microbiota in prediction of hypertension. Conclusion: These findings represent specific alterations in microbial diversity, genes, species and functions of the hypertensive gut microbiome. Further studies on the causality relationship between hypertension and gut microbiota will offer new prospects for treating and preventing the hypertension and its associated diseases.
Keywords: gut microbiome; hypertension; metagenome-wide association study; microbial dysbiosis.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous