

Repositioning drugs for traumatic brain injury - N-acetyl cysteine and Phenserine
- PMID: 28886718
- PMCID: PMC5591517
- DOI: 10.1186/s12929-017-0377-1
Repositioning drugs for traumatic brain injury - N-acetyl cysteine and Phenserine
Abstract
Traumatic brain injury (TBI) is one of the most common causes of morbidity and mortality of both young adults of less than 45 years of age and the elderly, and contributes to about 30% of all injury deaths in the United States of America. Whereas there has been a significant improvement in our understanding of the mechanism that underpin the primary and secondary stages of damage associated with a TBI incident, to date however, this knowledge has not translated into the development of effective new pharmacological TBI treatment strategies. Prior experimental and clinical studies of drugs working via a single mechanism only may have failed to address the full range of pathologies that lead to the neuronal loss and cognitive impairment evident in TBI and other disorders. The present review focuses on two drugs with the potential to benefit multiple pathways considered important in TBI. Notably, both agents have already been developed into human studies for other conditions, and thus have the potential to be rapidly repositioned as TBI therapies. The first is N-acetyl cysteine (NAC) that is currently used in over the counter medications for its anti-inflammatory properties. The second is (-)-phenserine ((-)-Phen) that was originally developed as an experimental Alzheimer's disease (AD) drug. We briefly review background information about TBI and subsequently review literature suggesting that NAC and (-)-Phen may be useful therapeutic approaches for TBI, for which there are no currently approved drugs.
Keywords: N-acetyl cysteine; Phenserine; Traumatic brain injury.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Robert E. Becker does not have a competing interest but a conflict of interest. For disclosure, Robert E. Becker holds a patent on the use of phenserine in concussion/TBI and Alzheimer’s disease assigned to Aristea Translational Medicine Corporation of Utah. None of the other authors have any competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
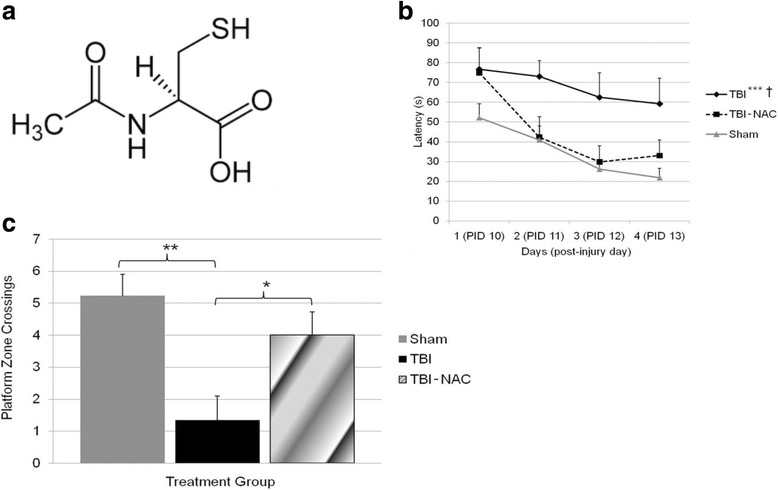
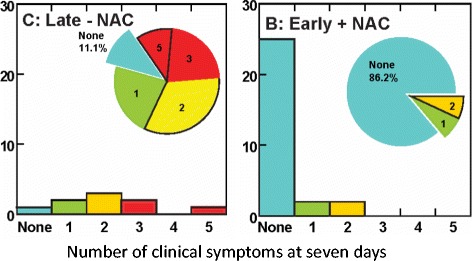
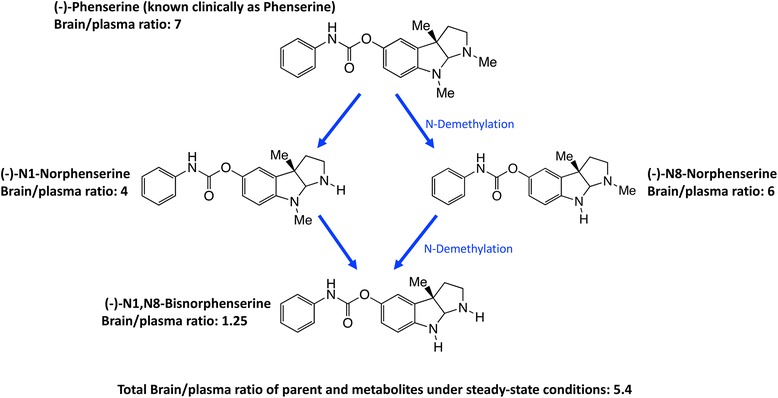
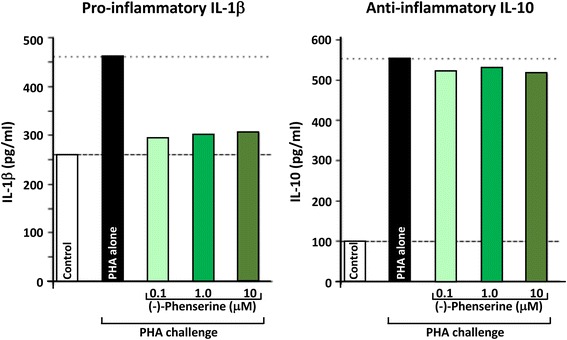
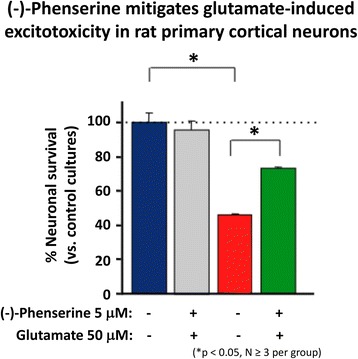
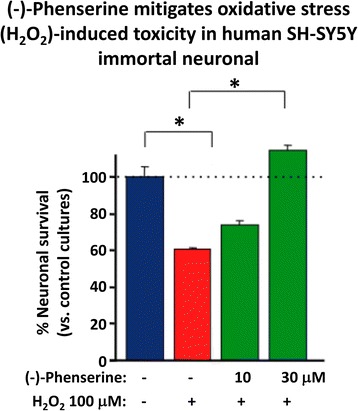
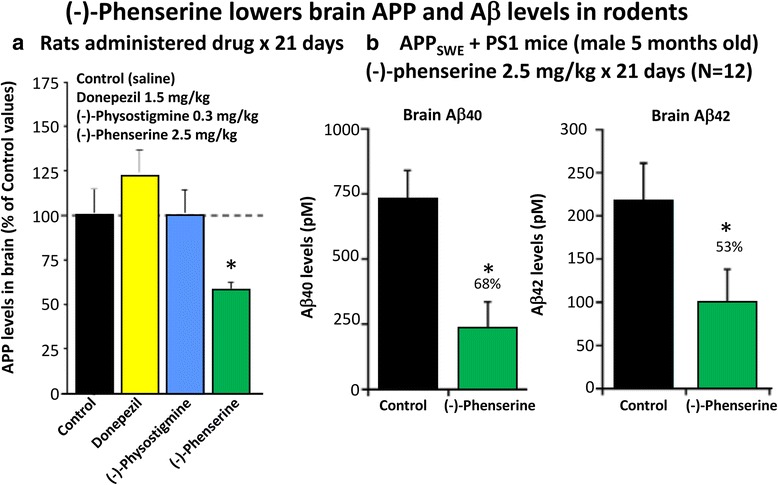
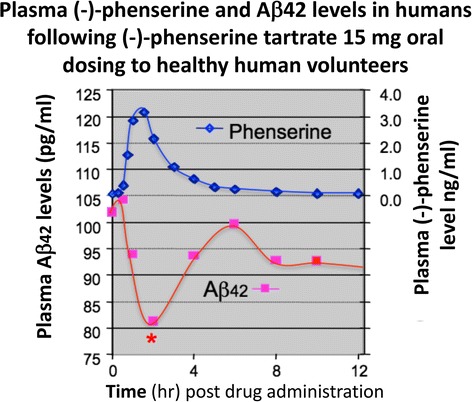
References
-
- Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Ko-busingye OC. The impact of traumatic brain injuries: a global perspective. Neuro Rehabilitation. 2007;22:341–353. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
