

Treating pediatric neuromuscular disorders: The future is now
- PMID: 28889642
- PMCID: PMC5900978
- DOI: 10.1002/ajmg.a.38418
Treating pediatric neuromuscular disorders: The future is now
Abstract
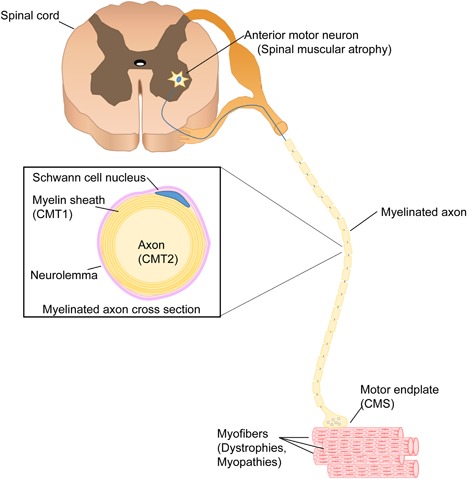
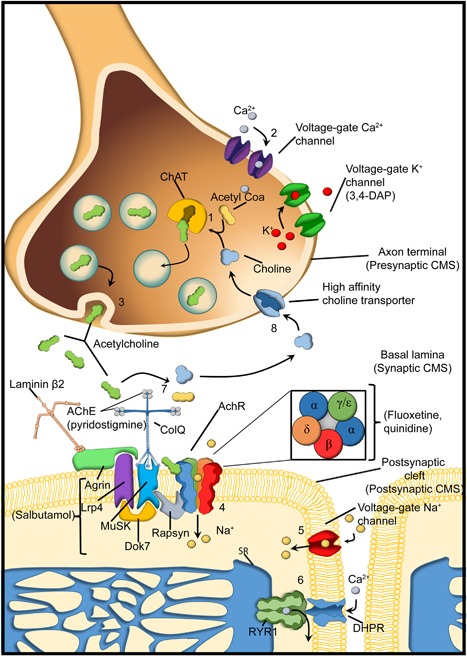
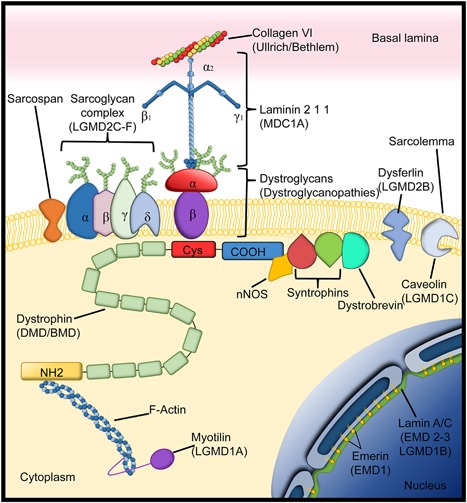
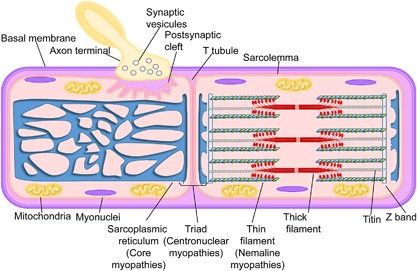
Pediatric neuromuscular diseases encompass all disorders with onset in childhood and where the primary area of pathology is in the peripheral nervous system. These conditions are largely genetic in etiology, and only those with a genetic underpinning will be presented in this review. This includes disorders of the anterior horn cell (e.g., spinal muscular atrophy), peripheral nerve (e.g., Charcot-Marie-Tooth disease), the neuromuscular junction (e.g., congenital myasthenic syndrome), and the muscle (myopathies and muscular dystrophies). Historically, pediatric neuromuscular disorders have uniformly been considered to be without treatment possibilities and to have dire prognoses. This perception has gradually changed, starting in part with the discovery and widespread application of corticosteroids for Duchenne muscular dystrophy. At present, several exciting therapeutic avenues are under investigation for a range of conditions, offering the potential for significant improvements in patient morbidities and mortality and, in some cases, curative intervention. In this review, we will present the current state of treatment for the most common pediatric neuromuscular conditions, and detail the treatment strategies with the greatest potential for helping with these devastating diseases.
Keywords: Charcot-Marie-Tooth disease; congenital myopathies; muscular dystrophies; neuromuscular disorders.
© 2017 The Authors. American Journal of Medical Genetics Part A Published by Wiley Periodicals, Inc.
Figures
References
-
- Abicht A., Muller J. S., Lochmuller H. (1993). Congenital Myasthenic syndromes. In R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E.Wallace, A. Amemiya, L. J. H. Bean, T. D. Bird, C. T. Fong, H. C. Mefford, R. J. H. Smith, & K. Stephens (Eds). Seattle WA: GeneReviews(R).
-
- Allen, H. D. , Flanigan, K. M. , Thrush, P. T. , Dvorchik, I. , Yin, H. , Canter, C. , … Mendell, J. R. (2013). A randomized, double‐blind trial of lisinopril and losartan for the treatment of cardiomyopathy in duchenne muscular dystrophy. PLoS Currents, 5, pii: ecurrents.md.2cc69a1dae4be7dfe2bcb420024ea865 https://doi.org/10.1371/currents.md.2cc69a1dae4be7dfe2bcb420024ea865 - DOI - PMC - PubMed
-
- Amthor, H. , & Hoogaars, W. M. (2012). Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Current Gene Therapy, 12(3), 245–259. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
