

Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance
- PMID: 28890882
- PMCID: PMC5574878
- DOI: 10.3389/fcimb.2017.00373
Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance
Abstract
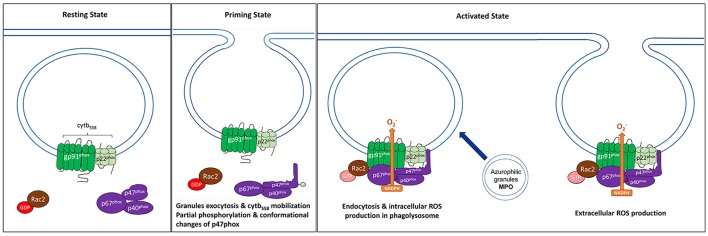
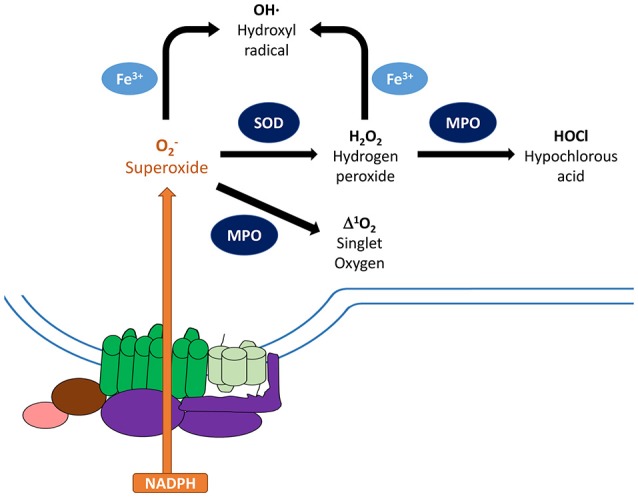
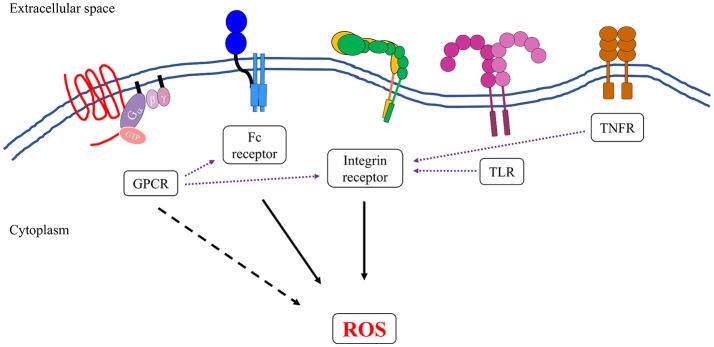
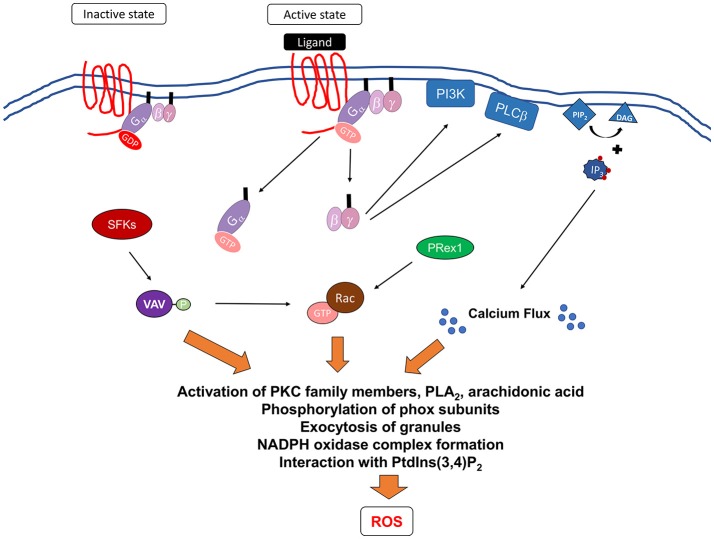
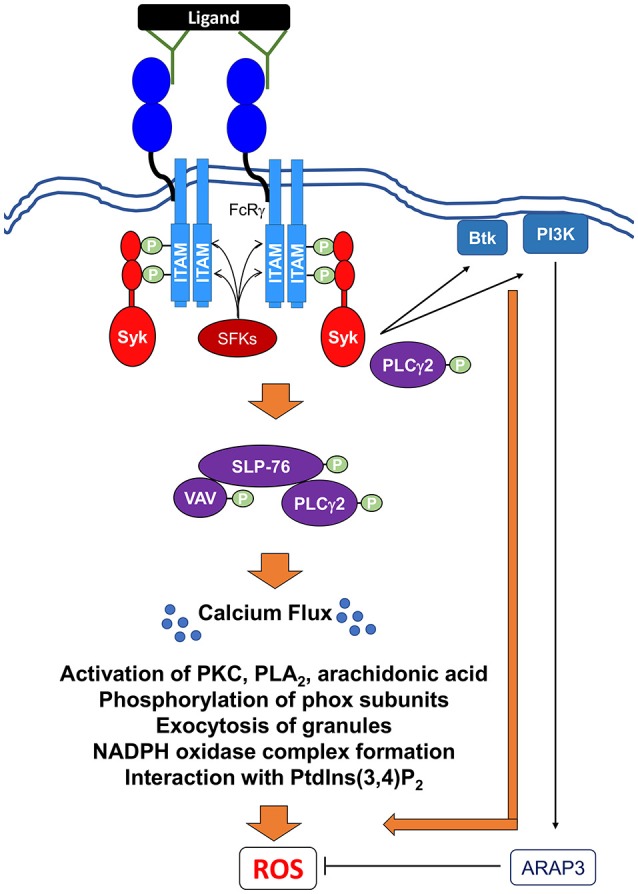
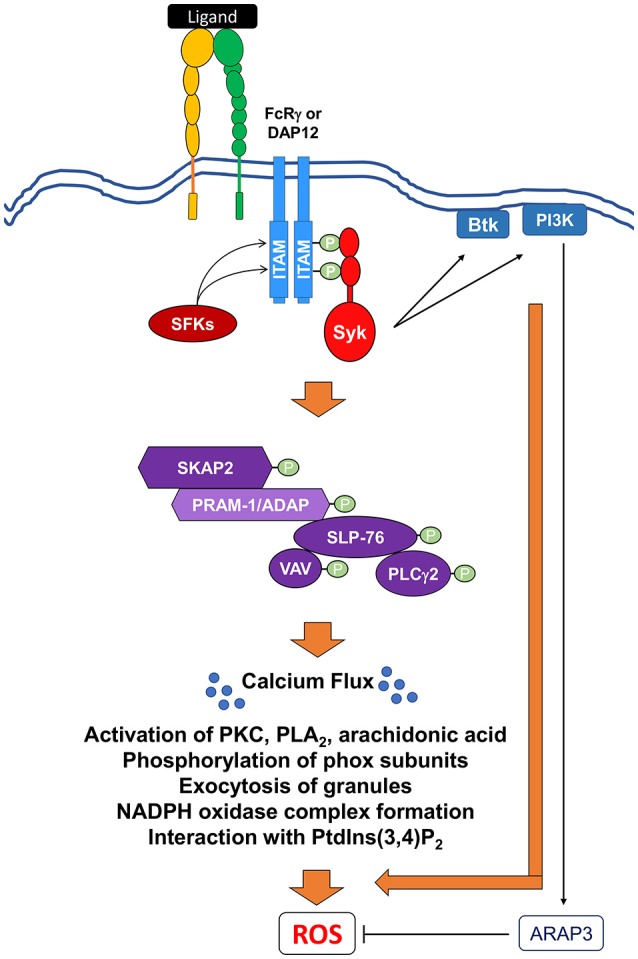
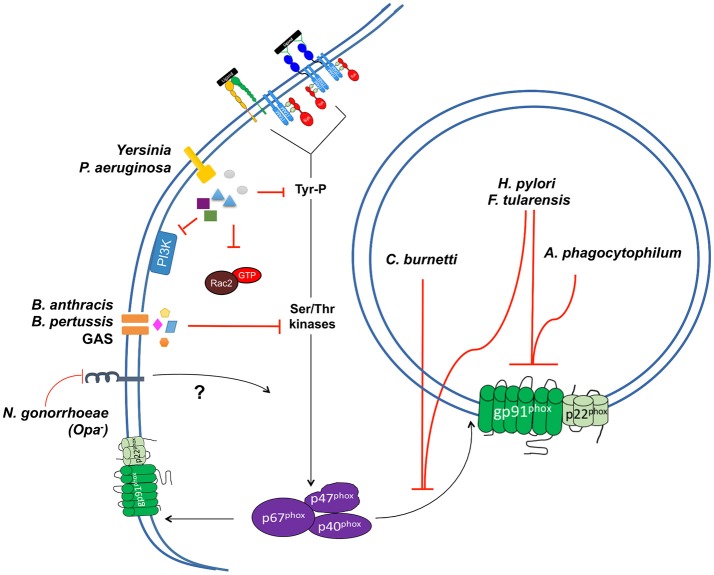
Reactive oxygen species (ROS) generated by NADPH oxidase play an important role in antimicrobial host defense and inflammation. Their deficiency in humans results in recurrent and severe bacterial infections, while their unregulated release leads to pathology from excessive inflammation. The release of high concentrations of ROS aids in clearance of invading bacteria. Localization of ROS release to phagosomes containing pathogens limits tissue damage. Host immune cells, like neutrophils, also known as PMNs, will release large amounts of ROS at the site of infection following the activation of surface receptors. The binding of ligands to G-protein-coupled receptors (GPCRs), toll-like receptors, and cytokine receptors can prime PMNs for a more robust response if additional signals are encountered. Meanwhile, activation of Fc and integrin directly induces high levels of ROS production. Additionally, GPCRs that bind to the bacterial-peptide analog fMLP, a neutrophil chemoattractant, can both prime cells and trigger low levels of ROS production. Engagement of these receptors initiates intracellular signaling pathways, resulting in activation of downstream effector proteins, assembly of the NADPH oxidase complex, and ultimately, the production of ROS by this complex. Within PMNs, ROS released by the NADPH oxidase complex can activate granular proteases and induce the formation of neutrophil extracellular traps (NETs). Additionally, ROS can cross the membranes of bacterial pathogens and damage their nucleic acids, proteins, and cell membranes. Consequently, in order to establish infections, bacterial pathogens employ various strategies to prevent restriction by PMN-derived ROS or downstream consequences of ROS production. Some pathogens are able to directly prevent the oxidative burst of phagocytes using secreted effector proteins or toxins that interfere with translocation of the NADPH oxidase complex or signaling pathways needed for its activation. Nonetheless, these pathogens often rely on repair and detoxifying proteins in addition to these secreted effectors and toxins in order to resist mammalian sources of ROS. This suggests that pathogens have both intrinsic and extrinsic mechanisms to avoid restriction by PMN-derived ROS. Here, we review mechanisms of oxidative burst in PMNs in response to bacterial infections, as well as the mechanisms by which bacterial pathogens thwart restriction by ROS to survive under conditions of oxidative stress.
Keywords: CGD; Fc receptors; G protein coupled receptors; NADPH oxidase; integrin receptors; neutrophils; reactive oxygen species; type 3 secreted effectors.
Figures
References
-
- Ago T., Nunoi H., Ito T., Sumimoto H. (1999). Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47(phox). Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47(phox), thereby activating the oxidase. J. Biol. Chem. 274, 33644–33653. - PubMed
-
- Ali H., Sozzani S., Fisher I., Barr A. J., Richardson R. M., Haribabu B., et al. (1998). Differential regulation of formyl peptide and platelet-activating factor receptors. Role of phospholipase Cbeta3 phosphorylation by protein kinase A. J. Biol. Chem. 273, 11012–11016. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical