

Phylotranscriptomic consolidation of the jawed vertebrate timetree
- PMID: 28890940
- PMCID: PMC5584656
- DOI: 10.1038/s41559-017-0240-5
Phylotranscriptomic consolidation of the jawed vertebrate timetree
Abstract
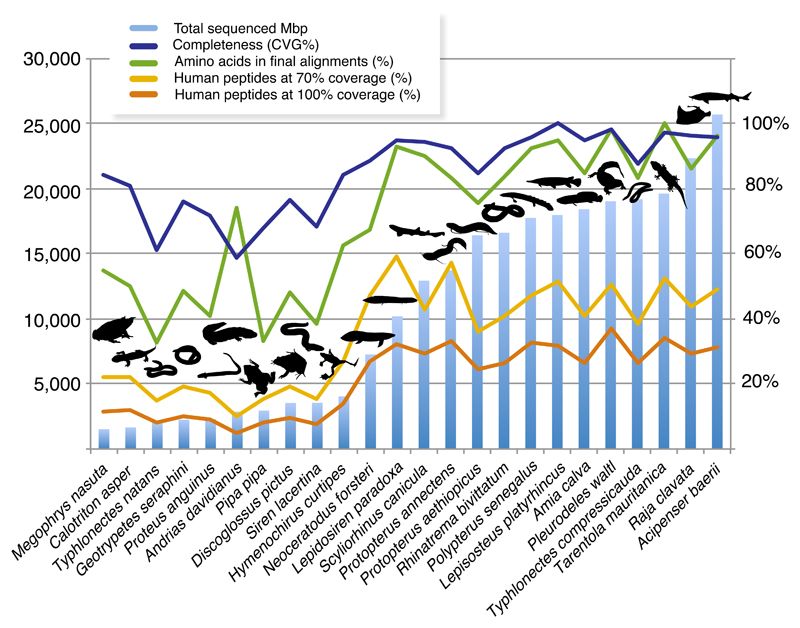
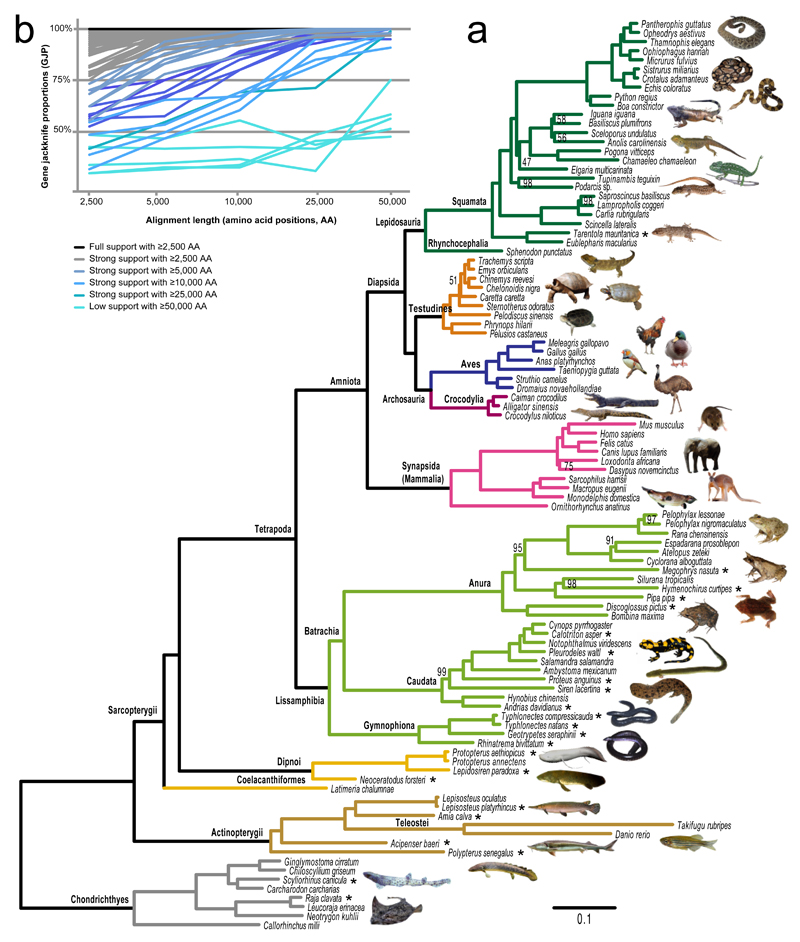
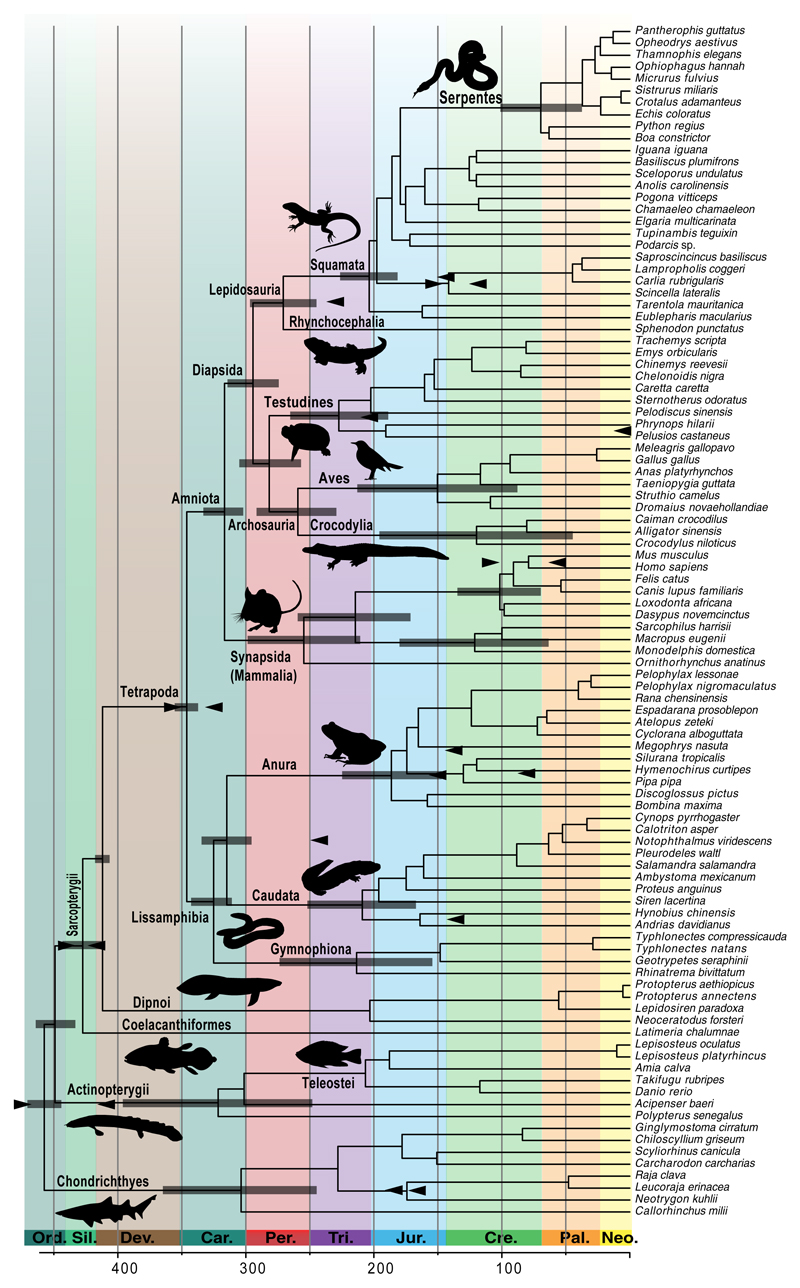
Phylogenomics is extremely powerful but introduces new challenges as no agreement exists on "standards" for data selection, curation and tree inference. We use jawed vertebrates (Gnathostomata) as model to address these issues. Despite considerable efforts in resolving their evolutionary history and macroevolution, few studies have included a full phylogenetic diversity of gnathostomes and some relationships remain controversial. We tested a novel bioinformatic pipeline to assemble large and accurate phylogenomic datasets from RNA sequencing and find this phylotranscriptomic approach successful and highly cost-effective. Increased sequencing effort up to ca. 10Gbp allows recovering more genes, but shallower sequencing (1.5Gbp) is sufficient to obtain thousands of full-length orthologous transcripts. We reconstruct a robust and strongly supported timetree of jawed vertebrates using 7,189 nuclear genes from 100 taxa, including 23 new transcriptomes from previously unsampled key species. Gene jackknifing of genomic data corroborates the robustness of our tree and allows calculating genome-wide divergence times by overcoming gene sampling bias. Mitochondrial genomes prove insufficient to resolve the deepest relationships because of limited signal and among-lineage rate heterogeneity. Our analyses emphasize the importance of large curated nuclear datasets to increase the accuracy of phylogenomics and provide a reference framework for the evolutionary history of jawed vertebrates.
Keywords: Gnathostomata; RNA-Seq; cross-validation; jackknifing; molecular dating; phylogeny; substitution rates; transcriptome.
Conflict of interest statement
Competing financial interests The authors declare no competing financial interests.
Figures
References
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
