

Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis
- PMID: 28891814
- PMCID: PMC5617658
- DOI: 10.1172/JCI94753
Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis
Abstract
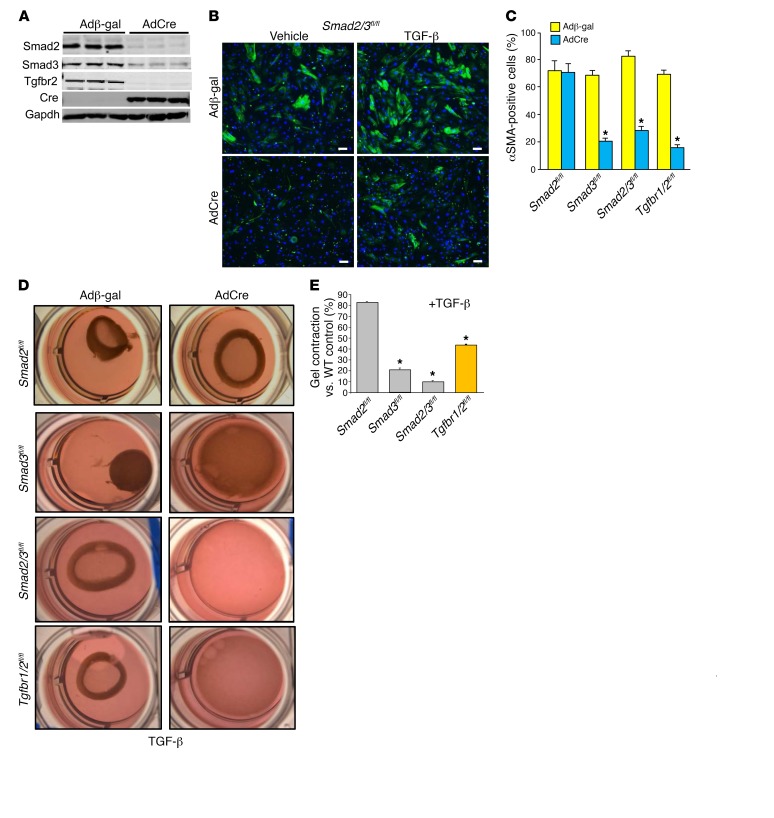
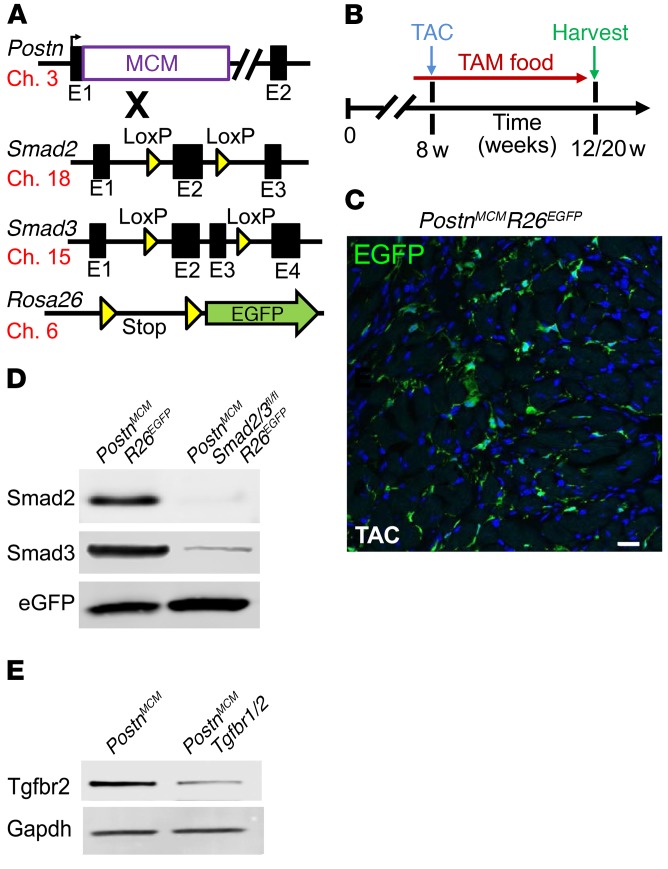
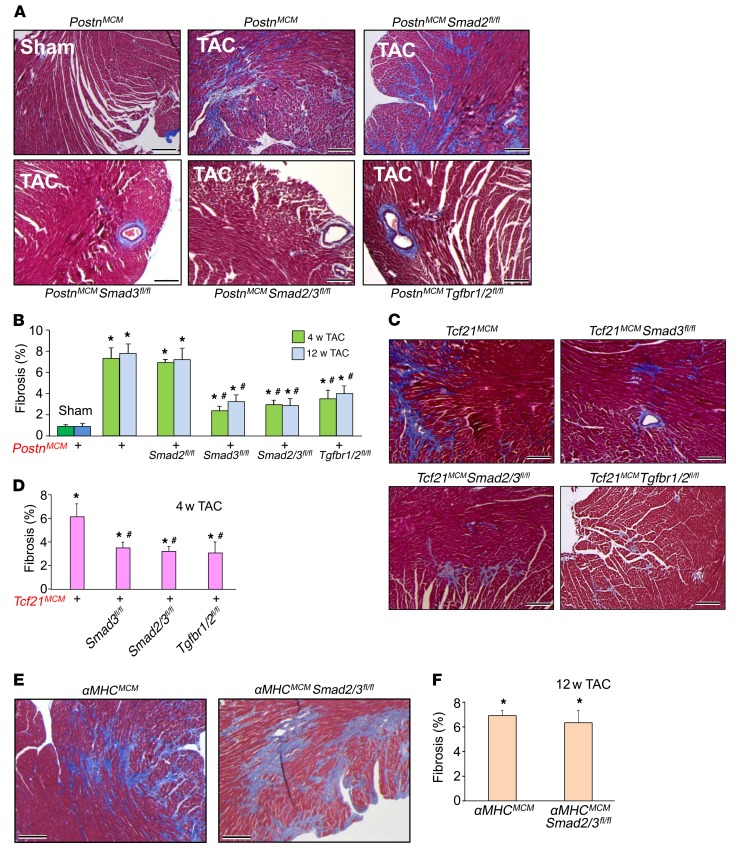
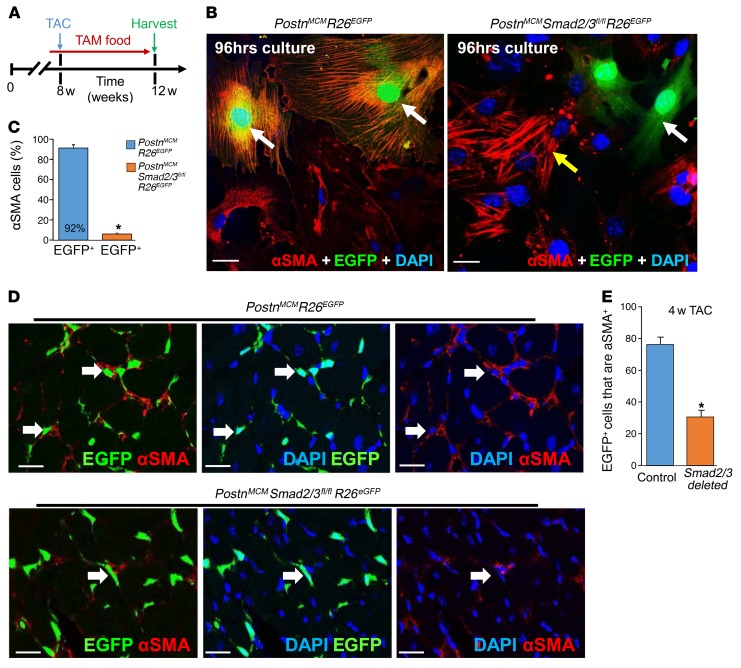
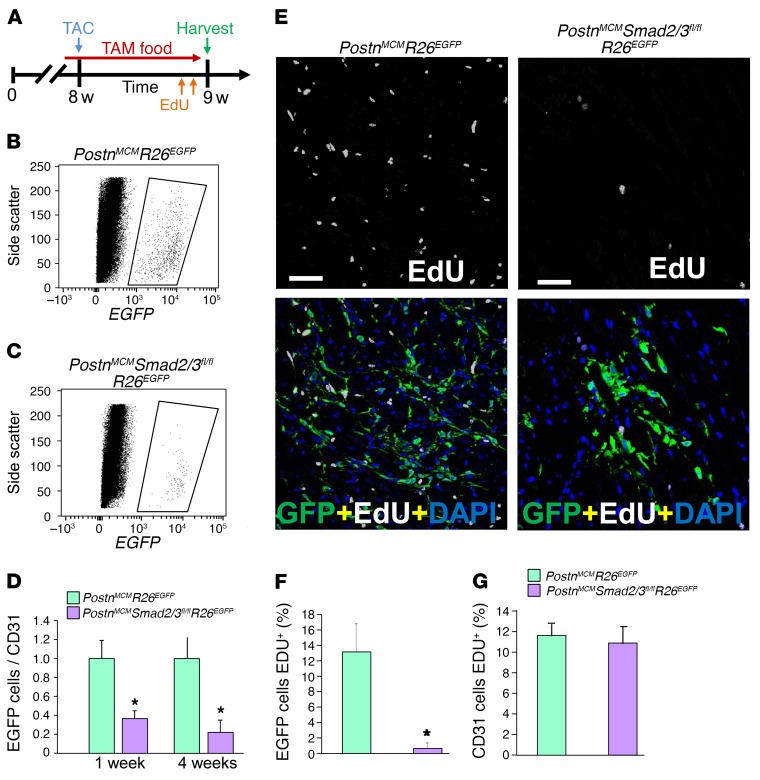
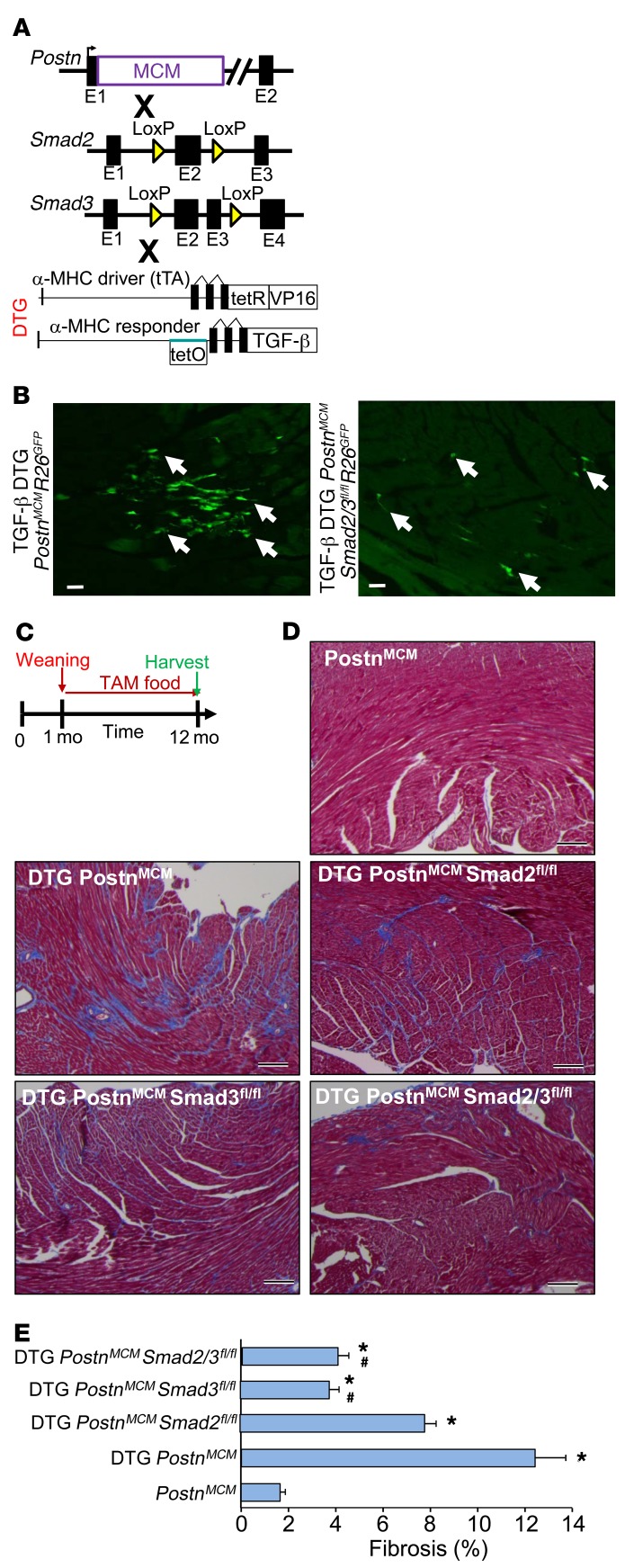
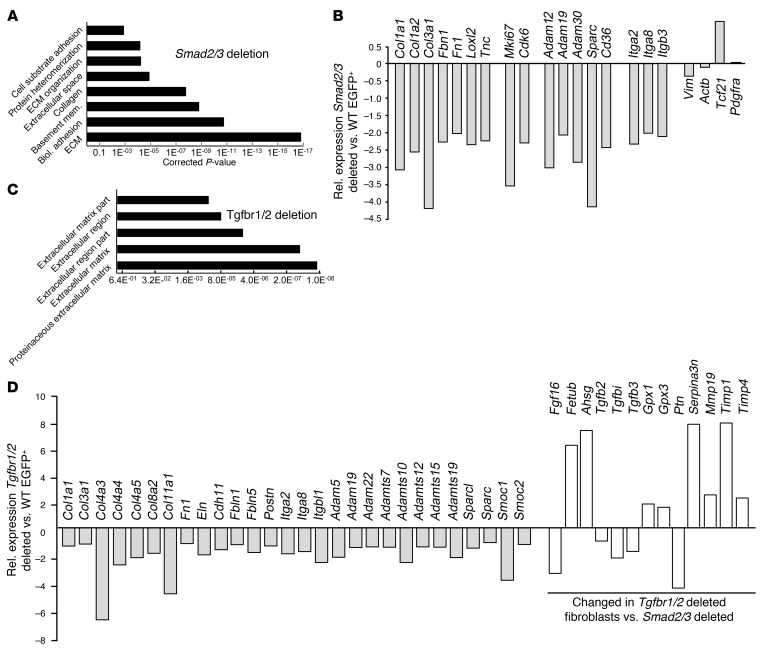
The master cytokine TGF-β mediates tissue fibrosis associated with inflammation and tissue injury. TGF-β induces fibroblast activation and differentiation into myofibroblasts that secrete extracellular matrix proteins. Canonical TGF-β signaling mobilizes Smad2 and Smad3 transcription factors that control fibrosis by promoting gene expression. However, the importance of TGF-β-Smad2/3 signaling in fibroblast-mediated cardiac fibrosis has not been directly evaluated in vivo. Here, we examined pressure overload-induced cardiac fibrosis in fibroblast- and myofibroblast-specific inducible Cre-expressing mouse lines with selective deletion of the TGF-β receptors Tgfbr1/2, Smad2, or Smad3. Fibroblast-specific deletion of Tgfbr1/2 or Smad3, but not Smad2, markedly reduced the pressure overload-induced fibrotic response as well as fibrosis mediated by a heart-specific, latency-resistant TGF-β mutant transgene. Interestingly, cardiac fibroblast-specific deletion of Tgfbr1/2, but not Smad2/3, attenuated the cardiac hypertrophic response to pressure overload stimulation. Mechanistically, loss of Smad2/3 from tissue-resident fibroblasts attenuated injury-induced cellular expansion within the heart and the expression of fibrosis-mediating genes. Deletion of Smad2/3 or Tgfbr1/2 from cardiac fibroblasts similarly inhibited the gene program for fibrosis and extracellular matrix remodeling, although deletion of Tgfbr1/2 uniquely altered expression of an array of regulatory genes involved in cardiomyocyte homeostasis and disease compensation. These findings implicate TGF-β-Smad2/3 signaling in activated tissue-resident cardiac fibroblasts as principal mediators of the fibrotic response.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
