

p53-Dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions
- PMID: 28893313
- PMCID: PMC5594515
- DOI: 10.1186/s13046-017-0596-z
p53-Dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions
Abstract
Background: As an important cellular stress sensor phosphoprotein p53 can trigger cell cycle arrest and apoptosis and regulate autophagy. The p53 activity mainly depends on its transactivating function, however, how p53 can select one or another biological outcome is still a matter of profound studies. Our previous findings indicate that switching cancer cells in high glucose (HG) impairs p53 apoptotic function and the transcription of target gene PUMA.
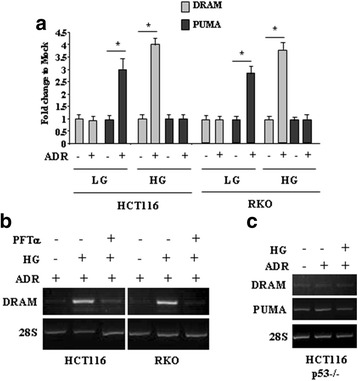
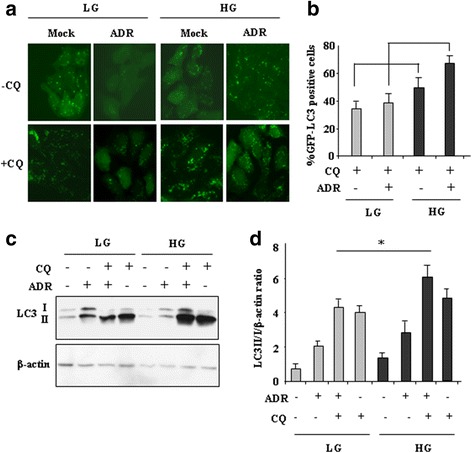
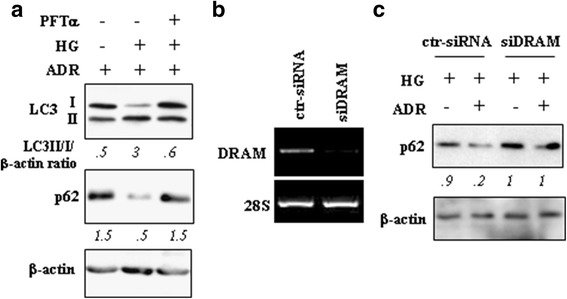
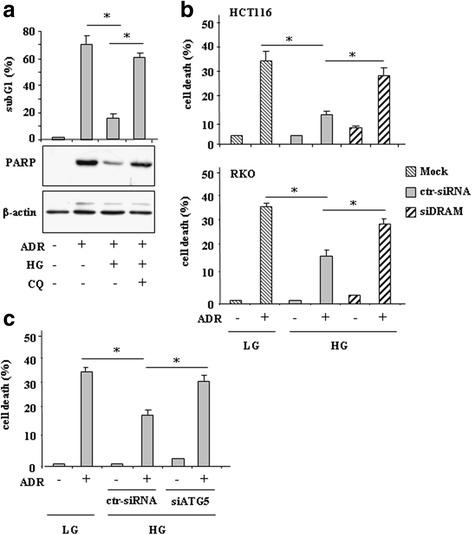
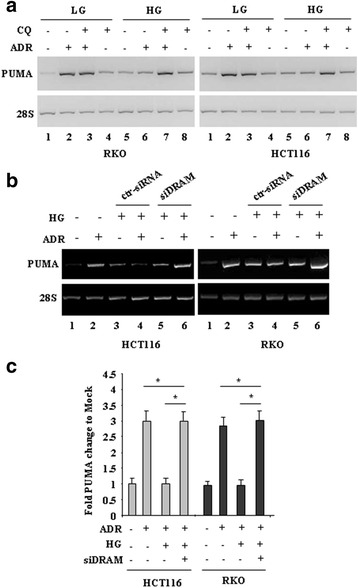
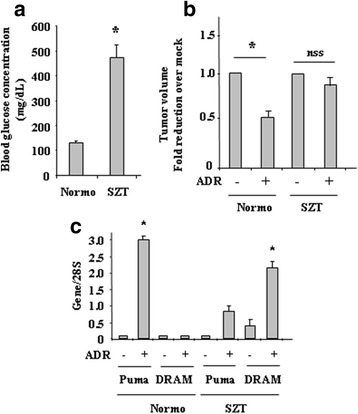
Methods and results: Here we report that, in response to drug adriamycin (ADR) in HG, p53 efficiently induced the expression of DRAM (damage-regulated autophagy modulator), a p53 target gene and a stress-induced regulator of autophagy. We found that ADR treatment of cancer cells in HG increased autophagy, as displayed by greater LC3II accumulation and p62 degradation compared to ADR-treated cells in low glucose. The increased autophagy in HG was in part dependent on p53-induced DRAM; indeed DRAM knockdown with specific siRNA reversed the expression of the autophagic markers in HG. A similar outcome was achieved by inhibiting p53 transcriptional activity with pifithrin-α. DRAM knockdown restored the ADR-induced cell death in HG to the levels obtained in low glucose. A similar outcome was achieved by inhibition of autophagy with cloroquine (CQ) or with silencing of autophagy gene ATG5. DRAM knockdown or inhibition of autophagy were both able to re-induce PUMA transcription in response to ADR, underlining a reciprocal interplay between PUMA to DRAM to unbalance p53 apoptotic activity in HG. Xenograft tumors transplanted in normoglycemic mice displayed growth delay after ADR treatment compared to those transplanted in diabetics mice and such different in vivo response correlated with PUMA to DRAM gene expression.
Conclusions: Altogether, these findings suggest that in normal/high glucose condition a mutual unbalance between p53-dependent apoptosis (PUMA) and autophagy (DRAM) gene occurred, modifying the ADR-induced cancer cell death in HG both in vitro and in vivo.
Keywords: Autophagy; Cancer; Chemotherapy; DRAM; Diabetes; Hyperglycemia; PUMA; p53.
Conflict of interest statement
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells.Oncol Rep. 2016 Jun;35(6):3639-47. doi: 10.3892/or.2016.4752. Epub 2016 Apr 19. Oncol Rep. 2016. PMID: 27109777
-
p53 Mediates Colistin-Induced Autophagy and Apoptosis in PC-12 Cells.Antimicrob Agents Chemother. 2016 Aug 22;60(9):5294-301. doi: 10.1128/AAC.00641-16. Print 2016 Sep. Antimicrob Agents Chemother. 2016. PMID: 27324771 Free PMC article.
-
Psammaplin A induces Sirtuin 1-dependent autophagic cell death in doxorubicin-resistant MCF-7/adr human breast cancer cells and xenografts.Biochim Biophys Acta. 2015 Feb;1850(2):401-10. doi: 10.1016/j.bbagen.2014.11.007. Epub 2014 Nov 12. Biochim Biophys Acta. 2015. PMID: 25445714
-
Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy.Autophagy. 2010 Jan;6(1):153-4. doi: 10.4161/auto.6.1.10537. Epub 2010 Jan 5. Autophagy. 2010. PMID: 19949306 Review.
-
TP53, TP53 Target Genes (DRAM, TIGAR), and Autophagy.Adv Exp Med Biol. 2019;1206:127-149. doi: 10.1007/978-981-15-0602-4_6. Adv Exp Med Biol. 2019. PMID: 31776983 Review.
Cited by
-
LC3-Associated Phagocytosis (LAP): A Potentially Influential Mediator of Efferocytosis-Related Tumor Progression and Aggressiveness.Front Oncol. 2020 Aug 5;10:1298. doi: 10.3389/fonc.2020.01298. eCollection 2020. Front Oncol. 2020. PMID: 32850405 Free PMC article. Review.
-
Lipid-induced DRAM recruits STOM to lysosomes and induces LMP to promote exosome release from hepatocytes in NAFLD.Sci Adv. 2021 Nov 5;7(45):eabh1541. doi: 10.1126/sciadv.abh1541. Epub 2021 Nov 3. Sci Adv. 2021. PMID: 34731006 Free PMC article.
-
Interplay between Endoplasmic Reticulum (ER) Stress and Autophagy Induces Mutant p53H273 Degradation.Biomolecules. 2020 Mar 3;10(3):392. doi: 10.3390/biom10030392. Biomolecules. 2020. PMID: 32138264 Free PMC article.
-
NRF2 in Cancer: Cross-Talk with Oncogenic Pathways and Involvement in Gammaherpesvirus-Driven Carcinogenesis.Int J Mol Sci. 2022 Dec 29;24(1):595. doi: 10.3390/ijms24010595. Int J Mol Sci. 2022. PMID: 36614036 Free PMC article. Review.
-
HIPK2 in Angiogenesis: A Promising Biomarker in Cancer Progression and in Angiogenic Diseases.Cancers (Basel). 2023 Mar 2;15(5):1566. doi: 10.3390/cancers15051566. Cancers (Basel). 2023. PMID: 36900356 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
