

Comparison of Single Genome and Allele Frequency Data Reveals Discordant Demographic Histories
- PMID: 28893846
- PMCID: PMC5677151
- DOI: 10.1534/g3.117.300259
Comparison of Single Genome and Allele Frequency Data Reveals Discordant Demographic Histories
Abstract
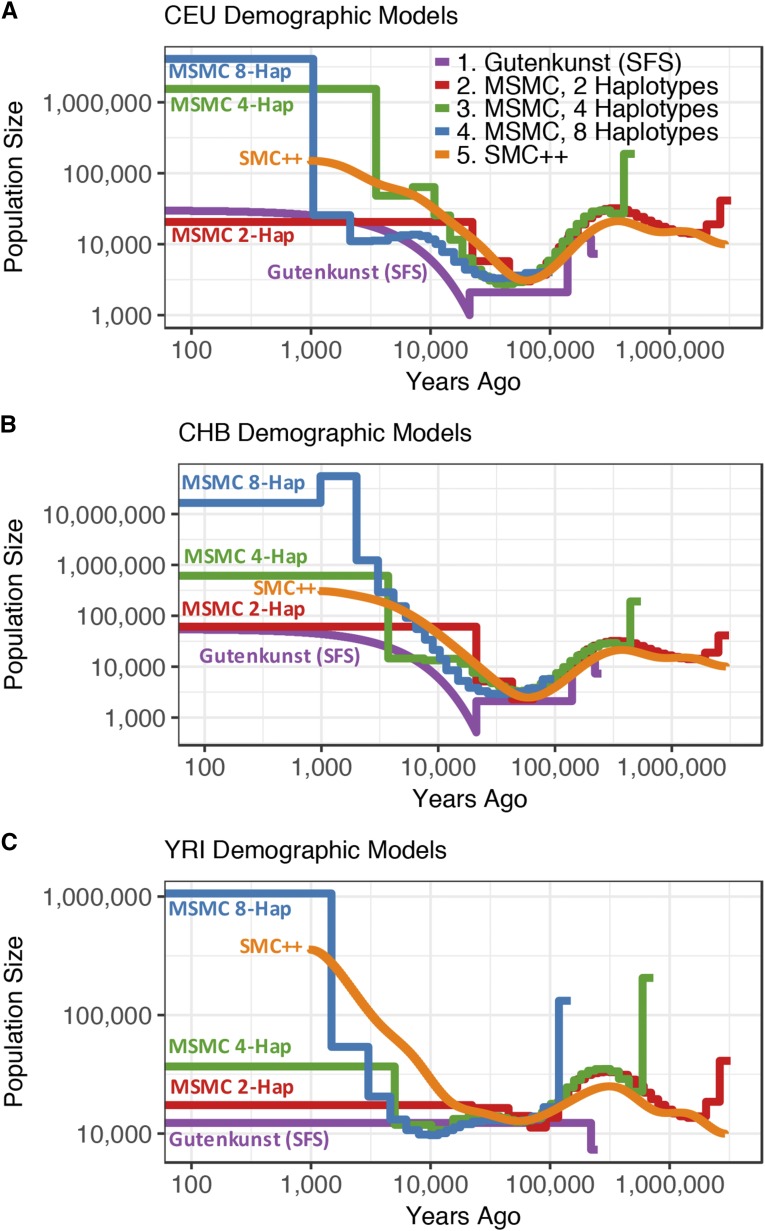
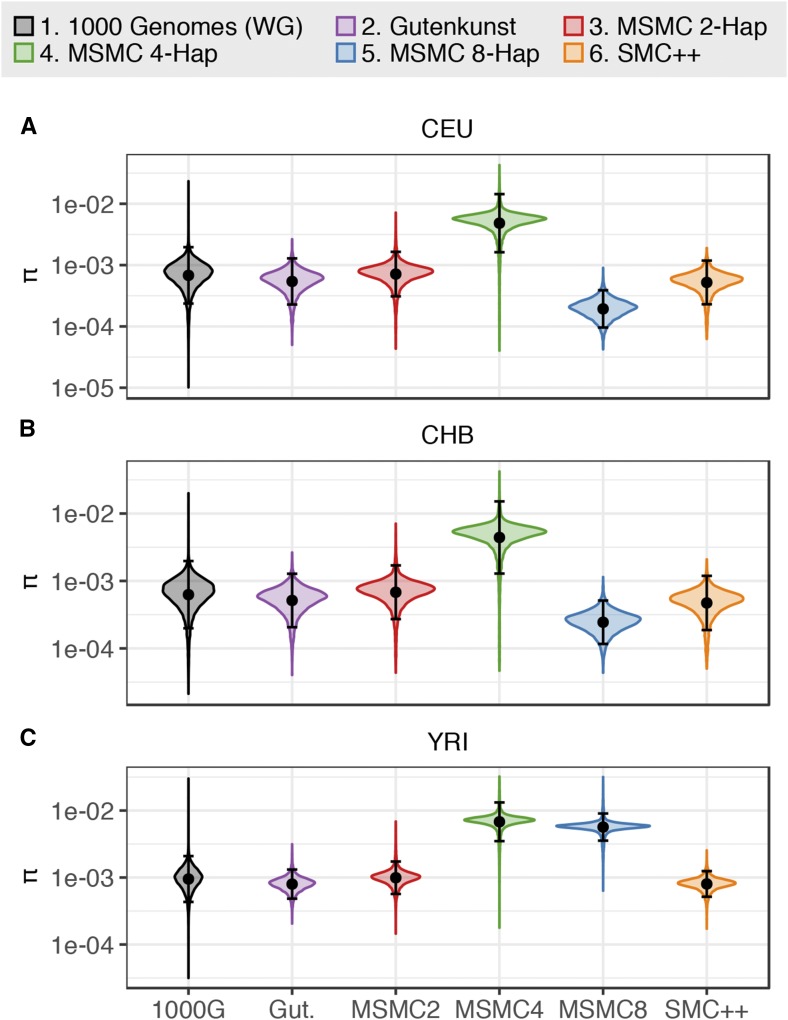
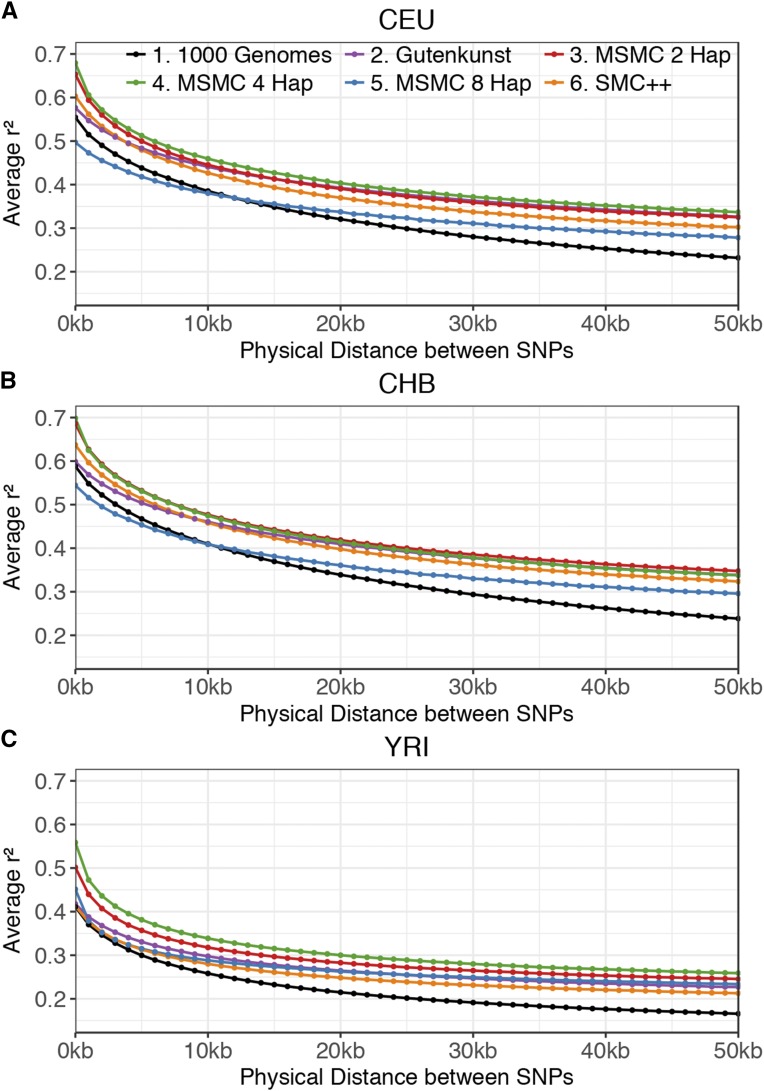
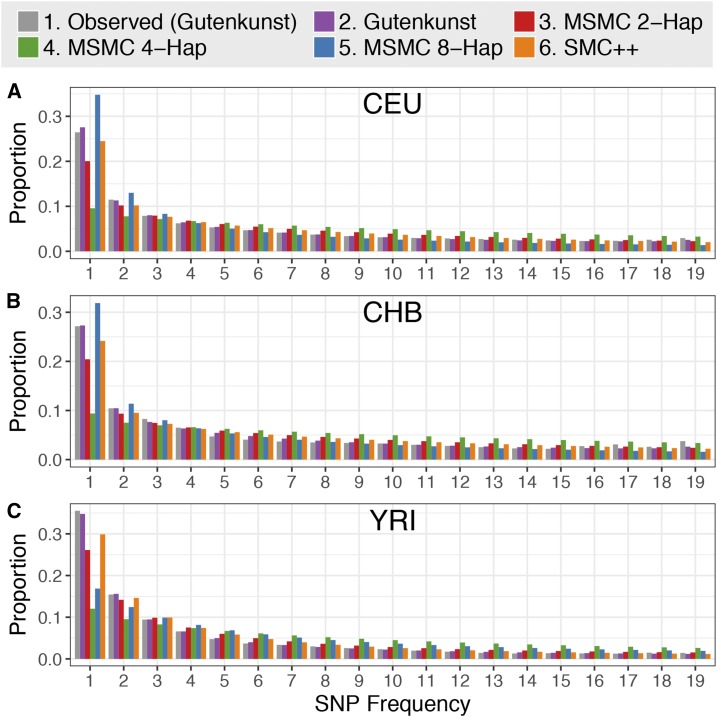
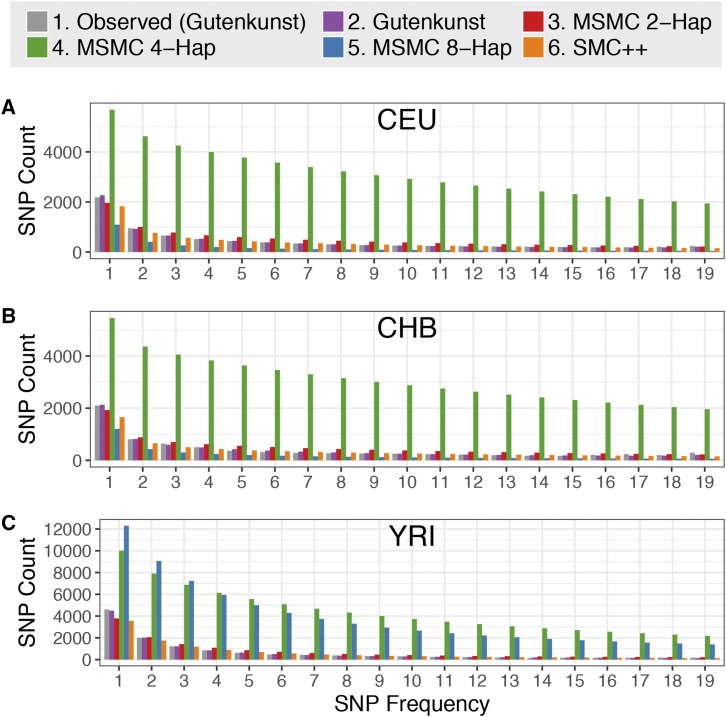
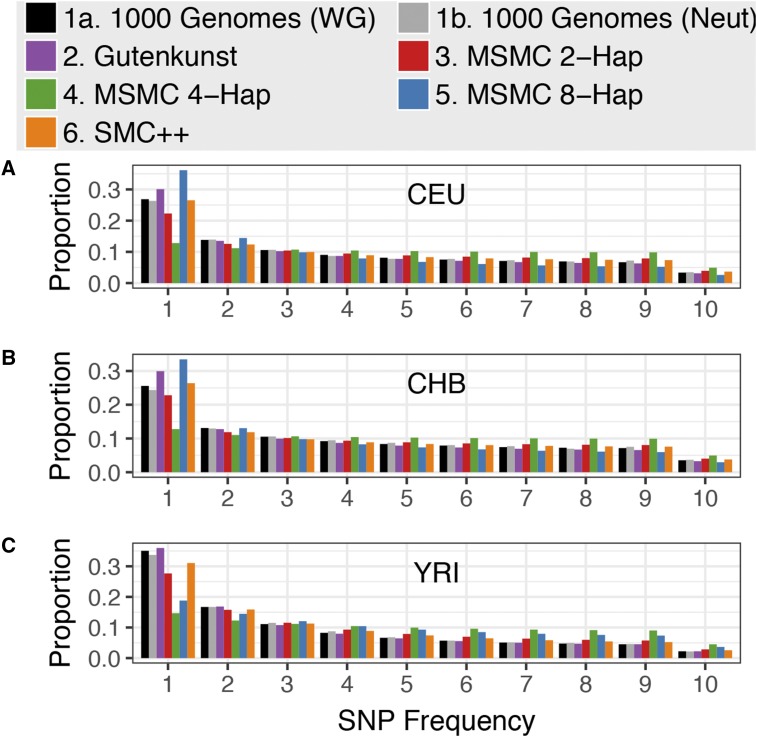
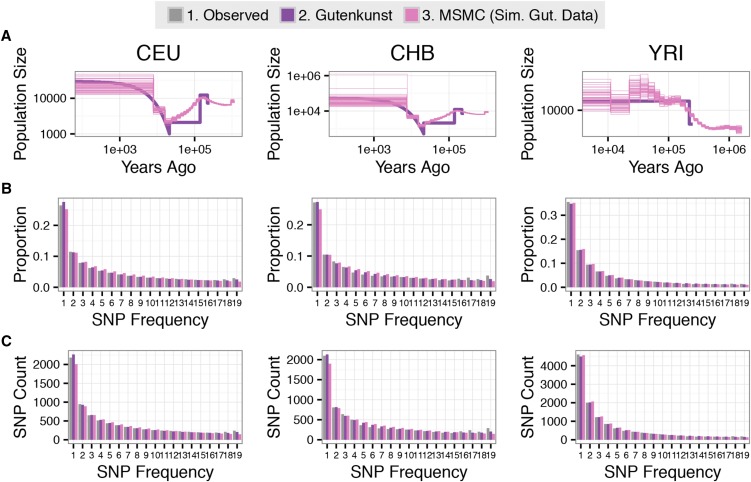
Inference of demographic history from genetic data is a primary goal of population genetics of model and nonmodel organisms. Whole genome-based approaches such as the pairwise/multiple sequentially Markovian coalescent methods use genomic data from one to four individuals to infer the demographic history of an entire population, while site frequency spectrum (SFS)-based methods use the distribution of allele frequencies in a sample to reconstruct the same historical events. Although both methods are extensively used in empirical studies and perform well on data simulated under simple models, there have been only limited comparisons of them in more complex and realistic settings. Here we use published demographic models based on data from three human populations (Yoruba, descendants of northwest-Europeans, and Han Chinese) as an empirical test case to study the behavior of both inference procedures. We find that several of the demographic histories inferred by the whole genome-based methods do not predict the genome-wide distribution of heterozygosity, nor do they predict the empirical SFS. However, using simulated data, we also find that the whole genome methods can reconstruct the complex demographic models inferred by SFS-based methods, suggesting that the discordant patterns of genetic variation are not attributable to a lack of statistical power, but may reflect unmodeled complexities in the underlying demography. More generally, our findings indicate that demographic inference from a small number of genomes, routine in genomic studies of nonmodel organisms, should be interpreted cautiously, as these models cannot recapitulate other summaries of the data.
Keywords: demographic inference; nonmodel organisms; pairwise sequentially Markovian coalescent; population genetics; site frequency spectrum.
Copyright © 2017 Beichman et al.
Figures
References
-
- Albert V. A., Barbazuk W. B., Der J. P., Leebens-Mack J., Ma H., et al. , 2013. The Amborella genome and the evolution of flowering plants. Science 342: 1241089. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources