

Heparan sulfate proteoglycans present PCSK9 to the LDL receptor
- PMID: 28894089
- PMCID: PMC5593881
- DOI: 10.1038/s41467-017-00568-7
Heparan sulfate proteoglycans present PCSK9 to the LDL receptor
Abstract
Coronary artery disease is the main cause of death worldwide and accelerated by increased plasma levels of cholesterol-rich low-density lipoprotein particles (LDL). Circulating PCSK9 contributes to coronary artery disease by inducing lysosomal degradation of the LDL receptor (LDLR) in the liver and thereby reducing LDL clearance. Here, we show that liver heparan sulfate proteoglycans are PCSK9 receptors and essential for PCSK9-induced LDLR degradation. The heparan sulfate-binding site is located in the PCSK9 prodomain and formed by surface-exposed basic residues interacting with trisulfated heparan sulfate disaccharide repeats. Accordingly, heparan sulfate mimetics and monoclonal antibodies directed against the heparan sulfate-binding site are potent PCSK9 inhibitors. We propose that heparan sulfate proteoglycans lining the hepatocyte surface capture PCSK9 and facilitates subsequent PCSK9:LDLR complex formation. Our findings provide new insights into LDL biology and show that targeting PCSK9 using heparan sulfate mimetics is a potential therapeutic strategy in coronary artery disease.PCSK9 interacts with LDL receptor, causing its degradation, and consequently reduces the clearance of LDL. Here, Gustafsen et al. show that PCSK9 interacts with heparan sulfate proteoglycans and this binding favors LDLR degradation. Pharmacological inhibition of this binding can be exploited as therapeutic intervention to lower LDL levels.
Conflict of interest statement
C.G., P.M., and S.G. are inventors on two patent applications on compounds for treating lipoprotein metabolism disorders submitted by Aarhus University. C.G., P.M., and S.G. have significant financial interest in Draupnir Bio ApS, a company that develops PCSK9 inhibitors and has exclusively licensed the above-mentioned intellectual property. The remaining authors declare no competing financial interests.
Figures
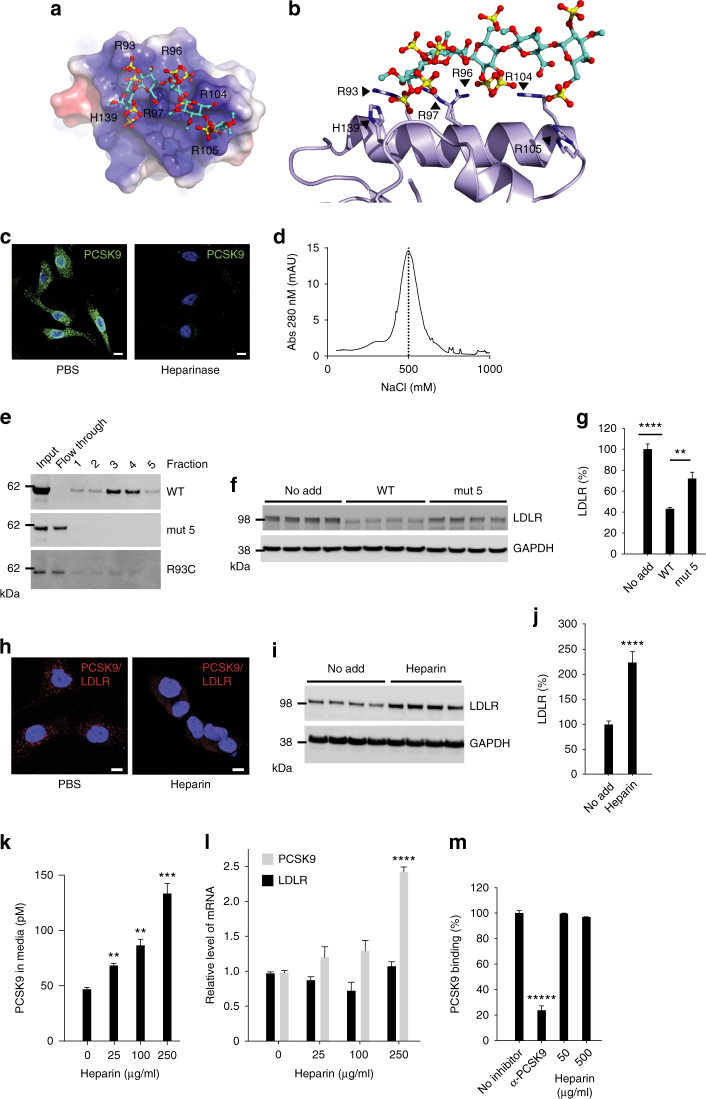
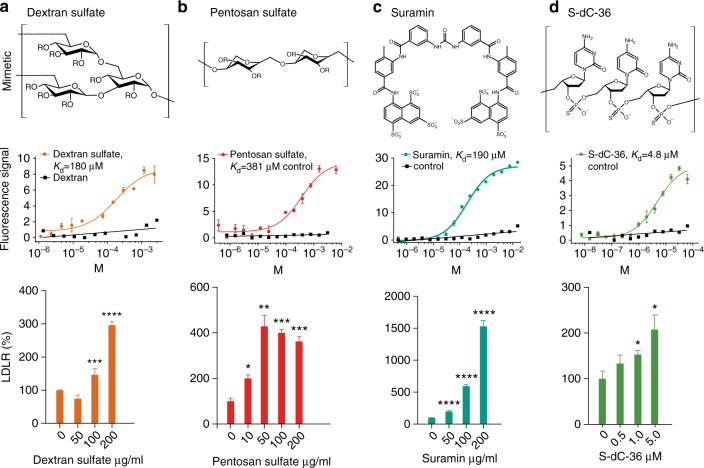
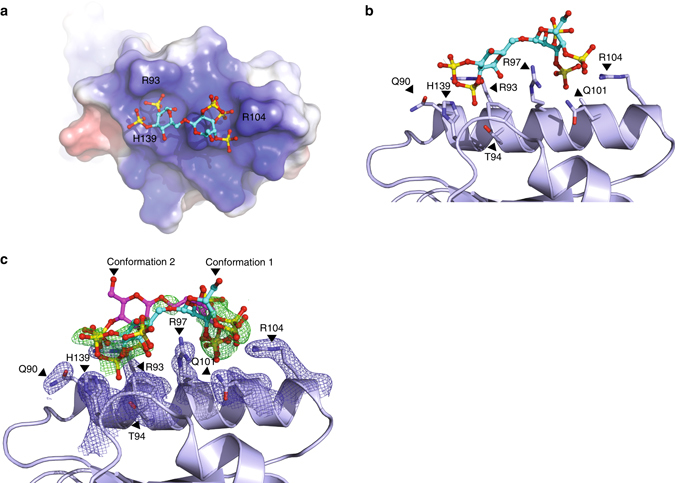
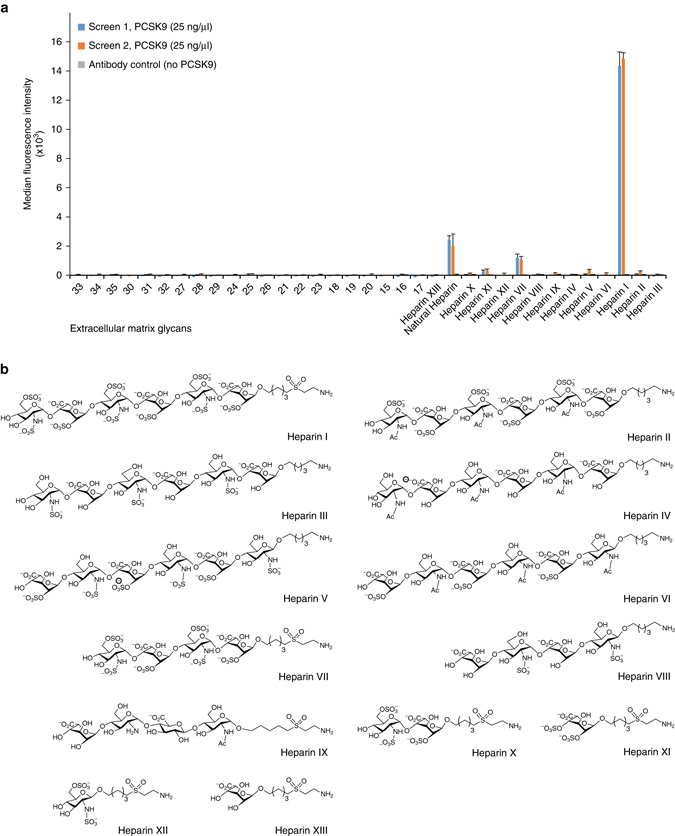
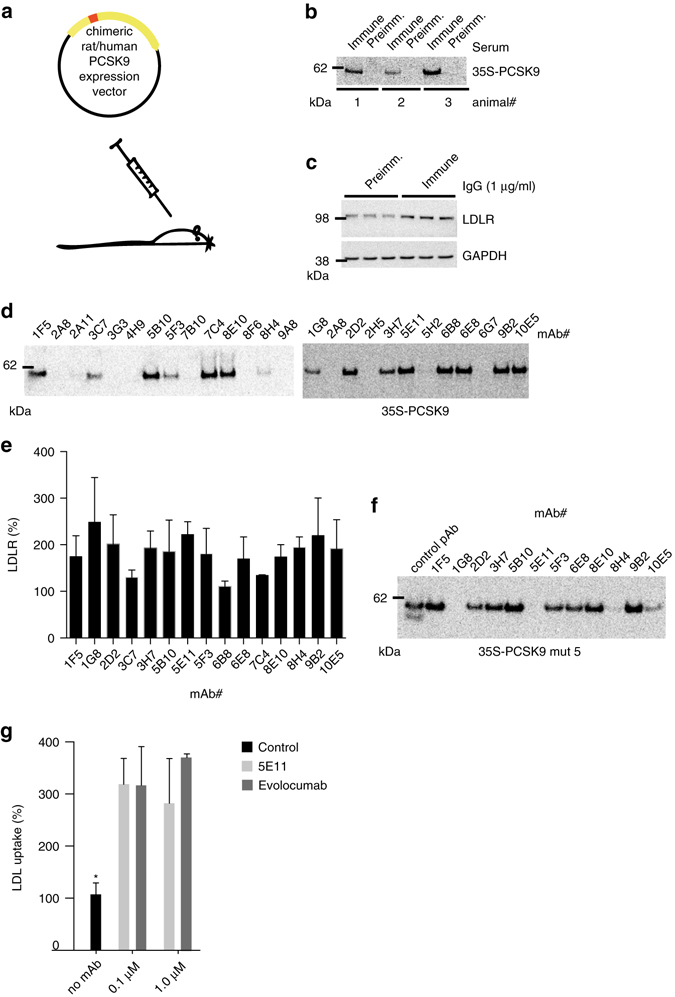
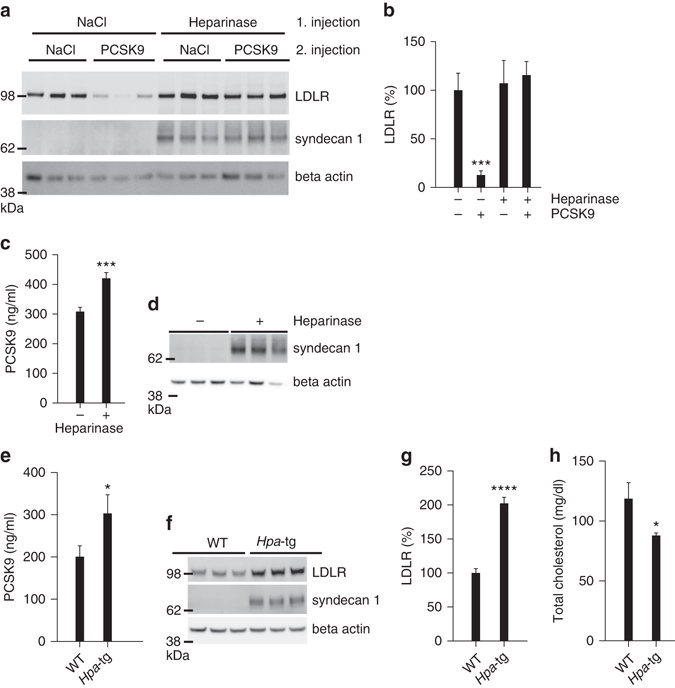
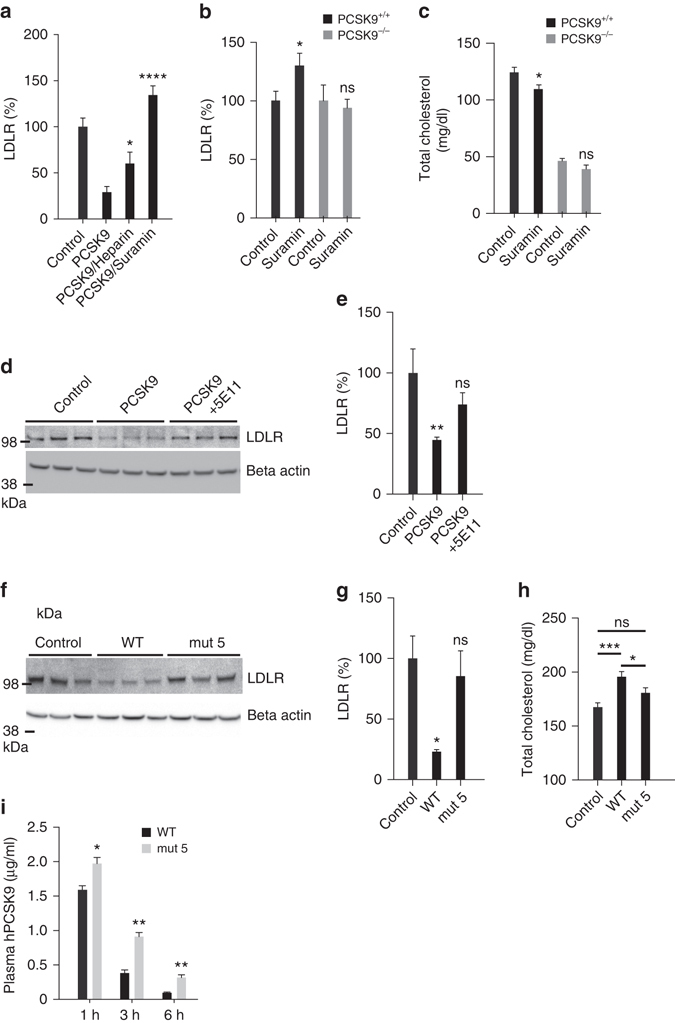
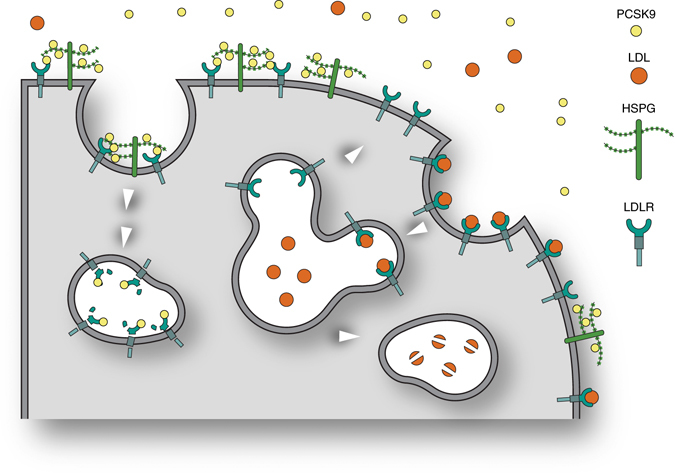
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
