

CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases
- PMID: 28895912
- PMCID: PMC5618314
- DOI: 10.3390/biomedicines5030056
CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases
Abstract
To date, no curative therapy is available for the treatment of most chronic inflammatory diseases such as atopic dermatitis, rheumatoid arthritis, or autoimmune disorders. Current treatments require a lifetime supply for patients to alleviate clinical symptoms and are unable to stop the course of disease. In contrast, a new series of immunotherapeutic agents targeting the Fc γ receptor I (CD64) have emerged and demonstrated significant clinical potential to actually resolving chronic inflammation driven by M1-type dysregulated macrophages. This subpopulation plays a key role in the initiation and maintenance of a series of chronic diseases. The novel recombinant M1-specific immunotherapeutics offer the prospect of highly effective treatment strategies as they have been shown to selectively eliminate the disease-causing macrophage subpopulations. In this review, we provide a detailed summary of the data generated, together with the advantages and the clinical potential of CD64-based targeted therapies for the treatment of chronic inflammatory diseases.
Keywords: CD64; chronic inflammatory disease; dysregulated macrophage; human cytolytic fusion protein; immunotherapy; immunotoxins.
Conflict of interest statement
Theo Thepen and Stefan Barth are co-inventors on CD64-related patents and patent applications, which are assigned to Fraunhofer or UCT. The authors otherwise declare no conflicts of interest.
Figures
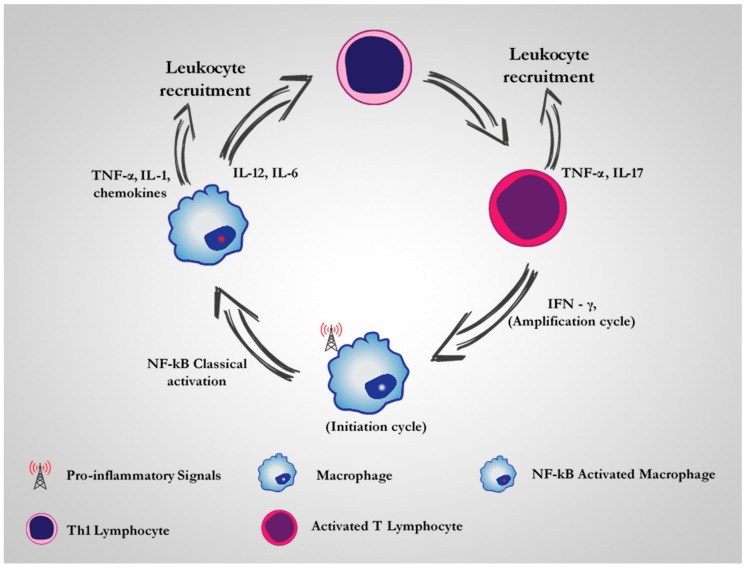
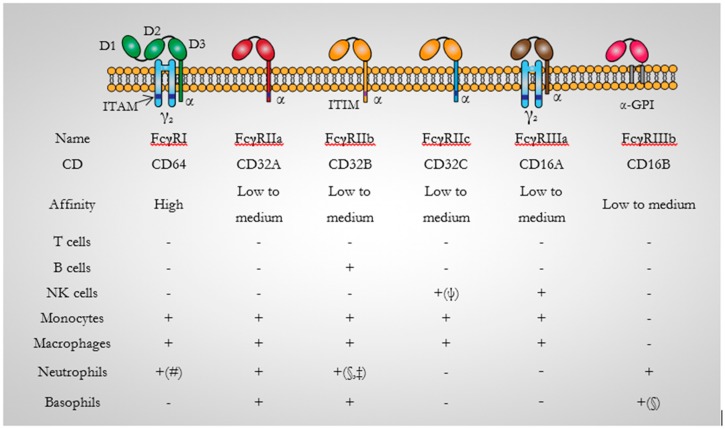
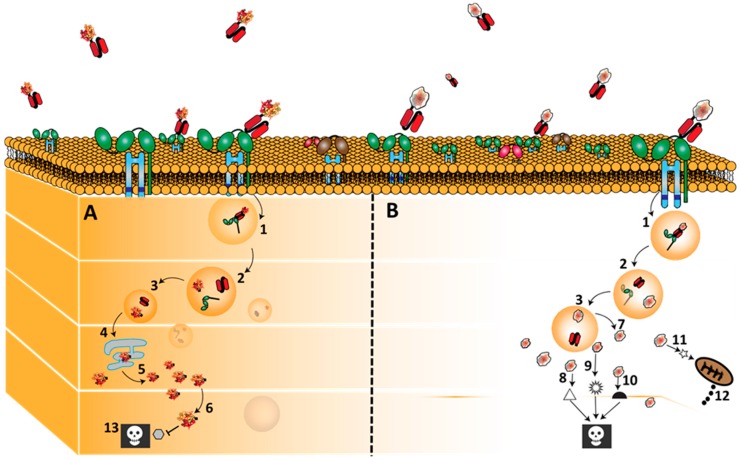
Similar articles
-
Macrophage-targeted therapy: CD64-based immunotoxins for treatment of chronic inflammatory diseases.Toxins (Basel). 2012 Sep;4(9):676-94. doi: 10.3390/toxins4090676. Epub 2012 Sep 14. Toxins (Basel). 2012. PMID: 23105975 Free PMC article. Review.
-
Targeting CD64 mediates elimination of M1 but not M2 macrophages in vitro and in cutaneous inflammation in mice and patient biopsies.MAbs. 2015;7(5):853-62. doi: 10.1080/19420862.2015.1066950. MAbs. 2015. PMID: 26218624 Free PMC article.
-
Recombinant H22(scFv) blocks CD64 and prevents the capture of anti-TNF monoclonal antibody. A potential strategy to enhance anti-TNF therapy.MAbs. 2014;6(5):1283-9. doi: 10.4161/mabs.32182. MAbs. 2014. PMID: 25517313 Free PMC article.
-
Recombinant, ETA'-based CD64 immunotoxins: improved efficacy by increased valency, both in vitro and in vivo in a chronic cutaneous inflammation model in human CD64 transgenic mice.Br J Dermatol. 2010 Aug;163(2):279-86. doi: 10.1111/j.1365-2133.2010.09824.x. Epub 2010 Apr 26. Br J Dermatol. 2010. PMID: 20426788
-
Fcgamma receptor 1 (CD64), a target beyond cancer.Curr Pharm Des. 2009;15(23):2712-8. doi: 10.2174/138161209788923967. Curr Pharm Des. 2009. PMID: 19689341 Review.
Cited by
-
NAAA-regulated lipid signaling in monocytes controls the induction of hyperalgesic priming in mice.Nat Commun. 2024 Feb 24;15(1):1705. doi: 10.1038/s41467-024-46139-5. Nat Commun. 2024. PMID: 38402219 Free PMC article.
-
Deep immunophenotyping reveals endometriosis is marked by dysregulation of the mononuclear phagocytic system in endometrium and peripheral blood.BMC Med. 2022 Apr 15;20(1):158. doi: 10.1186/s12916-022-02359-4. BMC Med. 2022. PMID: 35421980 Free PMC article.
-
Shifting Paradigms in Allergic Contact Dermatitis: The Role of Innate Immunity.J Invest Dermatol. 2020 Jan;140(1):21-28. doi: 10.1016/j.jid.2019.03.1133. Epub 2019 May 14. J Invest Dermatol. 2020. PMID: 31101475 Free PMC article. Review.
-
Abatacept downregulates Fcγ receptor I on circulating monocytes: a potential therapeutic mechanism in patients with rheumatoid arthritis.Arthritis Res Ther. 2022 Aug 13;24(1):194. doi: 10.1186/s13075-022-02886-8. Arthritis Res Ther. 2022. PMID: 35964055 Free PMC article.
-
Spatial visualization provides insight into immune modulation by an L-DBF vaccine formulation against Shigella.Front Immunol. 2025 Apr 23;16:1577040. doi: 10.3389/fimmu.2025.1577040. eCollection 2025. Front Immunol. 2025. PMID: 40336950 Free PMC article.
References
-
- Jain N.K., Mishra V., Mehra N.K. Targeted drug delivery to macrophages. Expert Opin. Drug Deliv. 2013;10:353–367. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
