

EMT and Treatment Resistance in Pancreatic Cancer
- PMID: 28895920
- PMCID: PMC5615337
- DOI: 10.3390/cancers9090122
EMT and Treatment Resistance in Pancreatic Cancer
Abstract
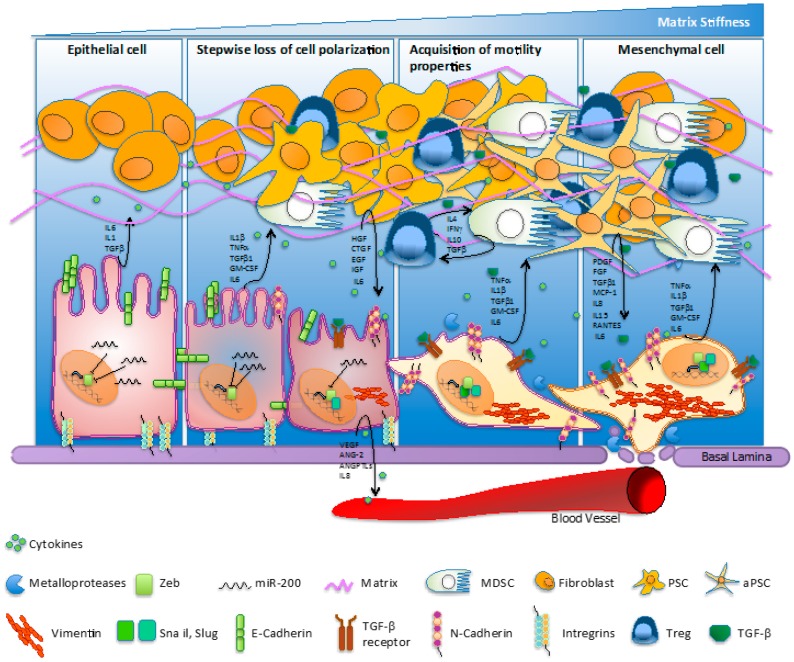
Pancreatic cancer (PC) is the third leading cause of adult cancer mortality in the United States. The poor prognosis for patients with PC is mainly due to its aggressive course, the limited efficacy of active systemic treatments, and a metastatic behavior, demonstrated throughout the evolution of the disease. On average, 80% of patients with PC are diagnosed with metastatic disease, and the half of those who undergo surgery and adjuvant therapy develop liver metastasis within two years. Metastatic dissemination is an early event in PC and is mainly attributed to an evolutionary biological process called epithelial-to-mesenchymal transition (EMT). This innate mechanism could have a dual role during embryonic growth and organ differentiation, and in cancer progression, cancer stem cell intravasation, and metastasis settlement. Many of the molecular pathways decisive in EMT progression have been already unraveled, but little is known about the causes behind the induction of this mechanism. EMT is one of the most distinctive and critical features of PC, occurring even in the very first stages of tumor development. This is known as pancreatic intraepithelial neoplasia (PanIN) and leads to early dissemination, drug resistance, and unfavorable prognosis and survival. The intention of this review is to shed new light on the critical role assumed by EMT during PC progression, with a particular focus on its role in PC resistance.
Keywords: EMT; pancreatic cancer; resistance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Epithelial-mesenchymal transition in pancreatic cancer: Is it a clinically significant factor?Biochim Biophys Acta. 2015 Jan;1855(1):43-9. doi: 10.1016/j.bbcan.2014.11.004. Epub 2014 Nov 26. Biochim Biophys Acta. 2015. PMID: 25432020 Review.
-
MicroRNA-204 attenuates the migration and invasion of pancreatic cancer cells by targeting ZEB1/EMT axis.Int J Clin Exp Pathol. 2018 Jul 1;11(7):3802-3811. eCollection 2018. Int J Clin Exp Pathol. 2018. PMID: 31949767 Free PMC article.
-
Epithelial Mesenchymal Transition in Aggressive Lung Cancers.Adv Exp Med Biol. 2016;890:37-56. doi: 10.1007/978-3-319-24932-2_3. Adv Exp Med Biol. 2016. PMID: 26703798 Review.
-
Overview of resistance to systemic therapy in patients with breast cancer.Adv Exp Med Biol. 2007;608:1-22. doi: 10.1007/978-0-387-74039-3_1. Adv Exp Med Biol. 2007. PMID: 17993229 Review.
-
Role of BCL9L in transforming growth factor-β (TGF-β)-induced epithelial-to-mesenchymal-transition (EMT) and metastasis of pancreatic cancer.Oncotarget. 2016 Nov 8;7(45):73725-73738. doi: 10.18632/oncotarget.12455. Oncotarget. 2016. PMID: 27713160 Free PMC article.
Cited by
-
Molecular Networks of Platinum Drugs and Their Interaction with microRNAs in Cancer.Genes (Basel). 2023 Nov 13;14(11):2073. doi: 10.3390/genes14112073. Genes (Basel). 2023. PMID: 38003016 Free PMC article.
-
Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions.Genome Med. 2020 Sep 29;12(1):80. doi: 10.1186/s13073-020-00776-9. Genome Med. 2020. PMID: 32988401 Free PMC article.
-
CPA4 Promotes EMT in Pancreatic Cancer via Stimulating PI3K-AKT-mTOR Signaling.Onco Targets Ther. 2020 Aug 25;13:8567-8580. doi: 10.2147/OTT.S257057. eCollection 2020. Onco Targets Ther. 2020. PMID: 32922037 Free PMC article.
-
Tumor Excision as a Metastatic Russian Roulette: Perioperative Interventions to Improve Long-Term Survival of Cancer Patients.Trends Cancer. 2020 Nov;6(11):951-959. doi: 10.1016/j.trecan.2020.06.004. Epub 2020 Jul 10. Trends Cancer. 2020. PMID: 32654993 Free PMC article. Review.
-
Mining the role of angiopoietin-like protein family in gastric cancer and seeking potential therapeutic targets by integrative bioinformatics analysis.Cancer Med. 2020 Jul;9(13):4850-4863. doi: 10.1002/cam4.3100. Epub 2020 May 14. Cancer Med. 2020. PMID: 32410376 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
