

A Dual Role of Caspase-8 in Triggering and Sensing Proliferation-Associated DNA Damage, a Key Determinant of Liver Cancer Development
- PMID: 28898696
- PMCID: PMC5598544
- DOI: 10.1016/j.ccell.2017.08.010
A Dual Role of Caspase-8 in Triggering and Sensing Proliferation-Associated DNA Damage, a Key Determinant of Liver Cancer Development
Abstract
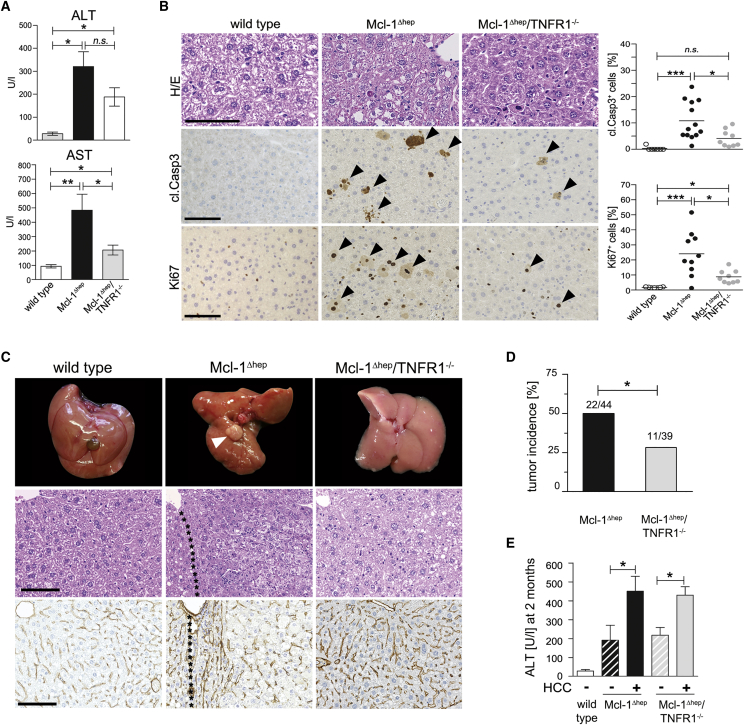
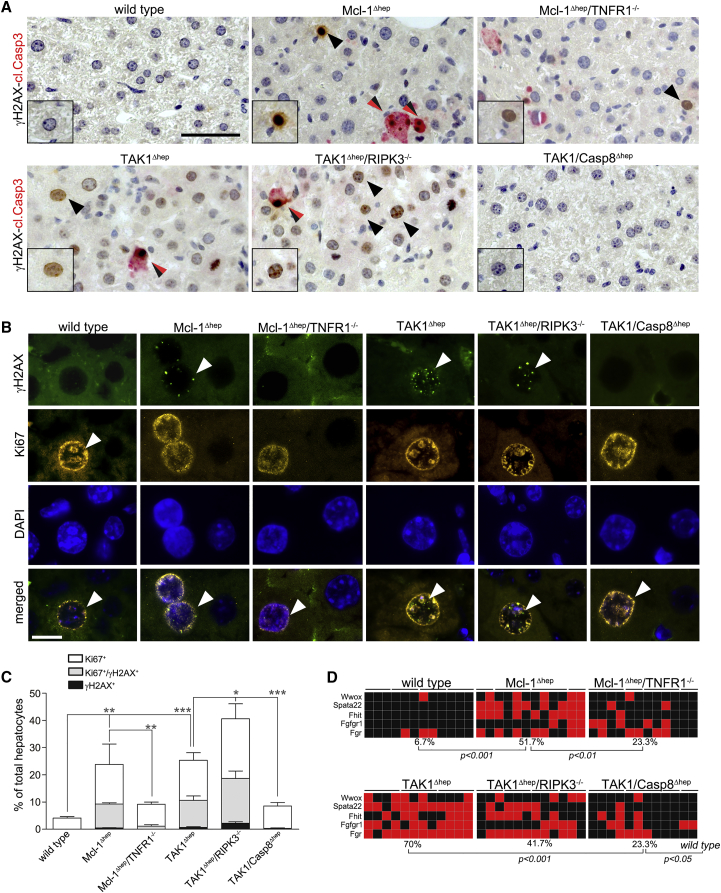
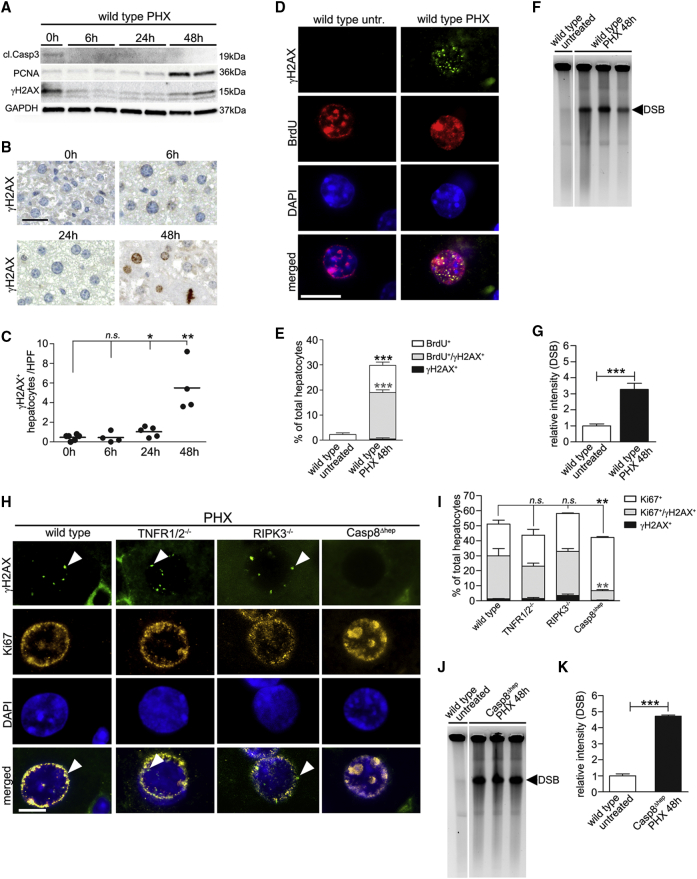
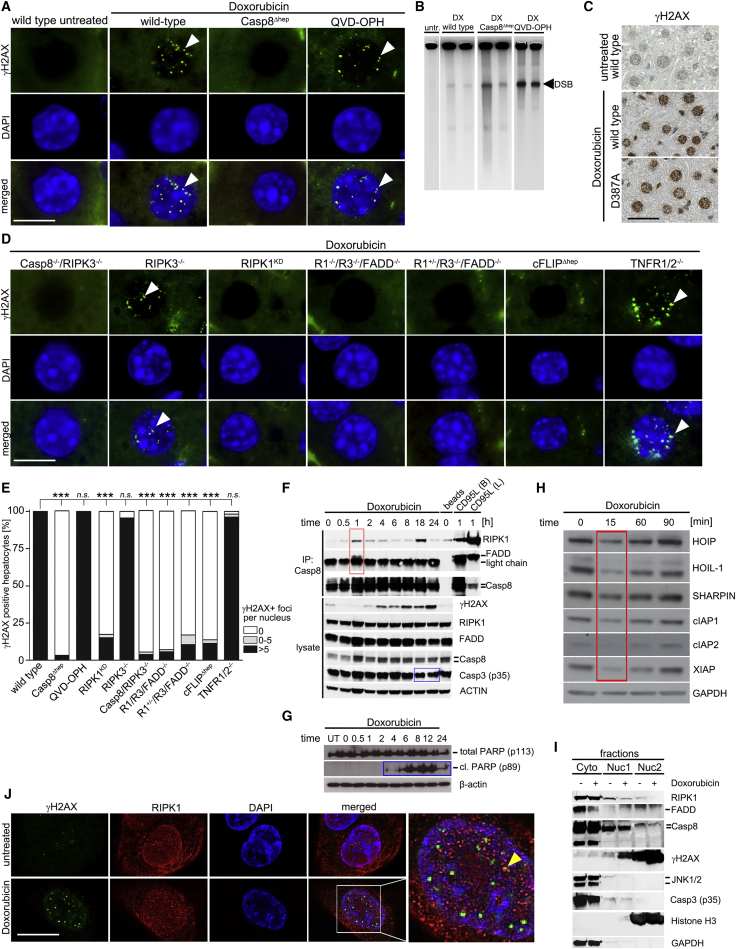
Concomitant hepatocyte apoptosis and regeneration is a hallmark of chronic liver diseases (CLDs) predisposing to hepatocellular carcinoma (HCC). Here, we mechanistically link caspase-8-dependent apoptosis to HCC development via proliferation- and replication-associated DNA damage. Proliferation-associated replication stress, DNA damage, and genetic instability are detectable in CLDs before any neoplastic changes occur. Accumulated levels of hepatocyte apoptosis determine and predict subsequent hepatocarcinogenesis. Proliferation-associated DNA damage is sensed by a complex comprising caspase-8, FADD, c-FLIP, and a kinase-dependent function of RIPK1. This platform requires a non-apoptotic function of caspase-8, but no caspase-3 or caspase-8 cleavage. It may represent a DNA damage-sensing mechanism in hepatocytes that can act via JNK and subsequent phosphorylation of the histone variant H2AX.
Keywords: DNA damage response; apoptosis; hepatocellular carcinoma; liver; replication stress.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures




References
-
- Affo S., Dominguez M., Lozano J.J., Sancho-Bru P., Rodrigo-Torres D., Morales-Ibanez O., Moreno M., Millan C., Loaeza-del-Castillo A., Altamirano J. Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut. 2013;62:452–460. - PMC - PubMed
-
- Alexiades M.R., Cepko C. Quantitative analysis of proliferation and cell cycle length during development of the rat retina. Dev. Dyn. 1996;205:293–307. - PubMed
-
- Batts K.P., Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am. J. Surg. Pathol. 1995;19:1409–1417. - PubMed
-
- Bettermann K., Vucur M., Haybaeck J., Koppe C., Janssen J., Heymann F., Weber A., Weiskirchen R., Liedtke C., Gassler N. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell. 2010;17:481–496. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
