

E46K α-synuclein pathological mutation causes cell-autonomous toxicity without altering protein turnover or aggregation
- PMID: 28900007
- PMCID: PMC5625897
- DOI: 10.1073/pnas.1703420114
E46K α-synuclein pathological mutation causes cell-autonomous toxicity without altering protein turnover or aggregation
Abstract
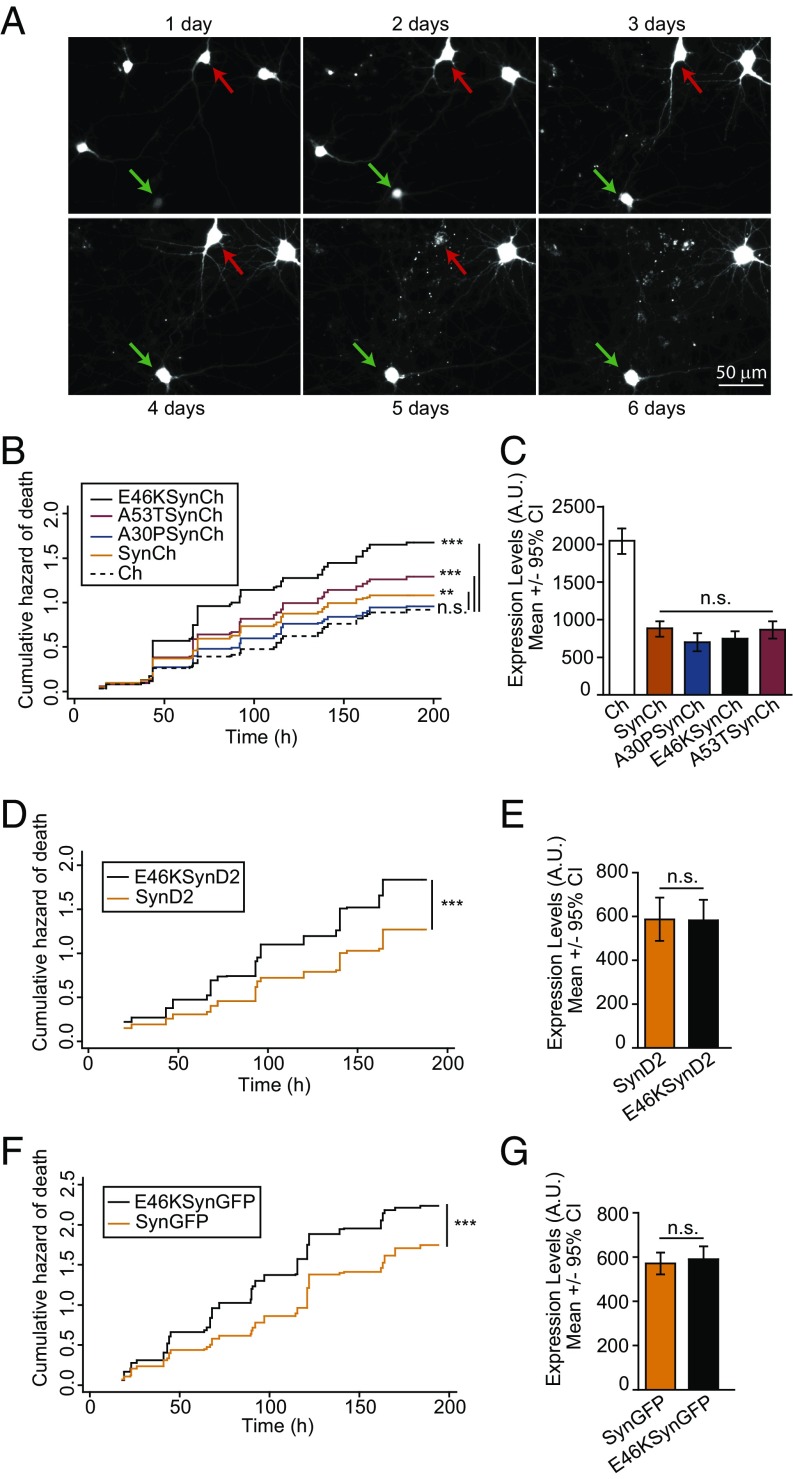
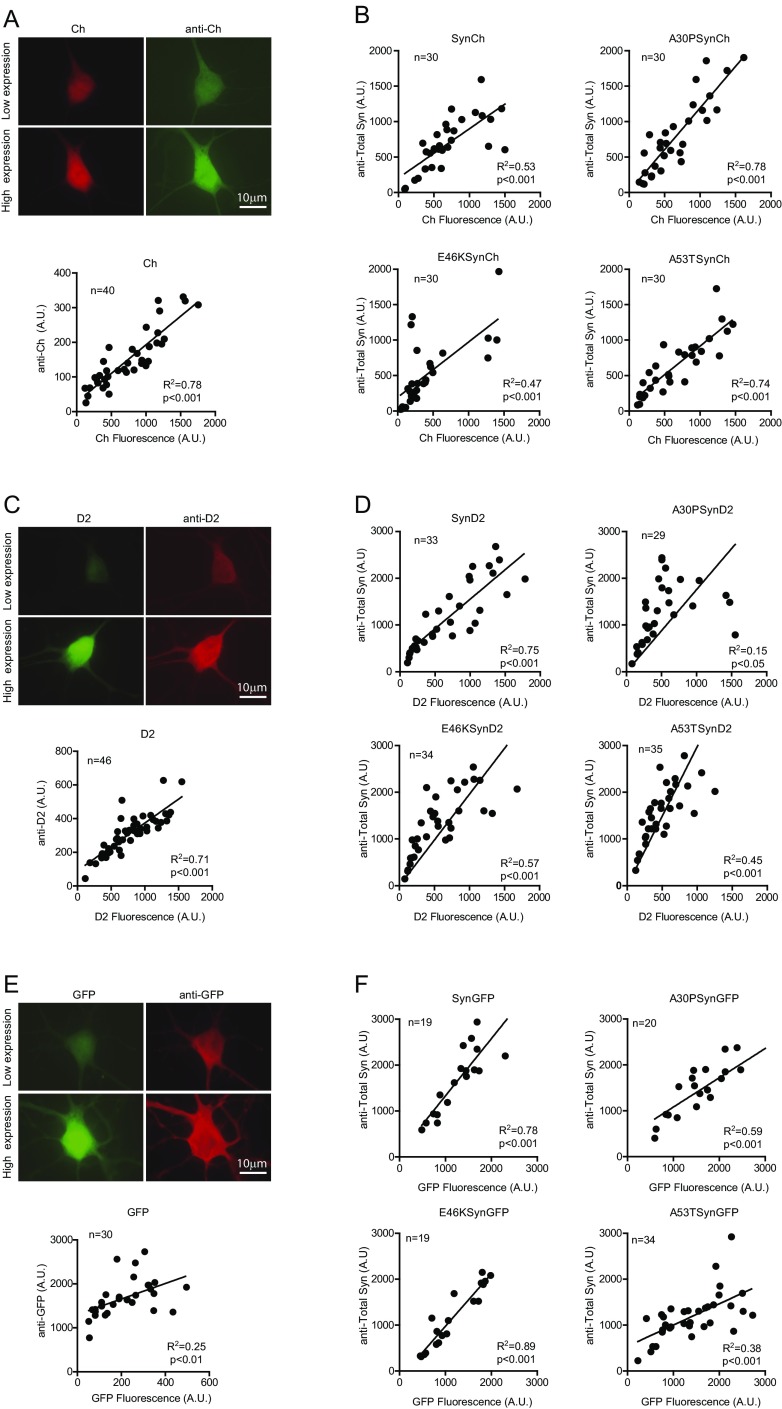
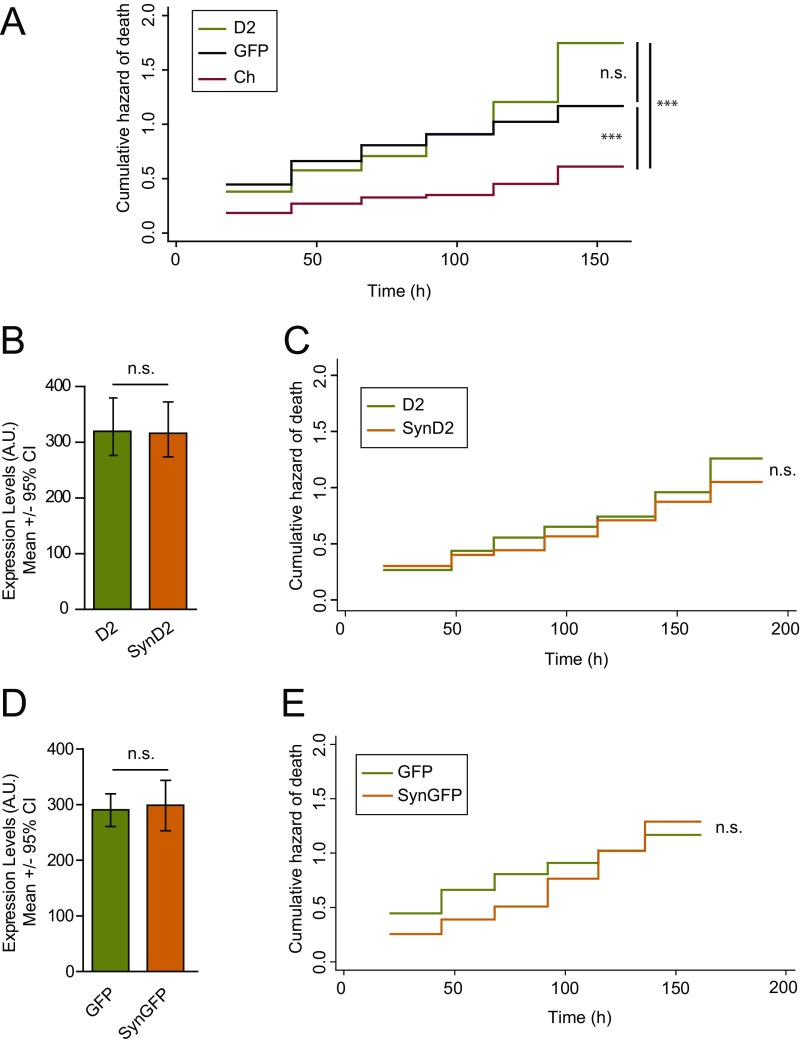
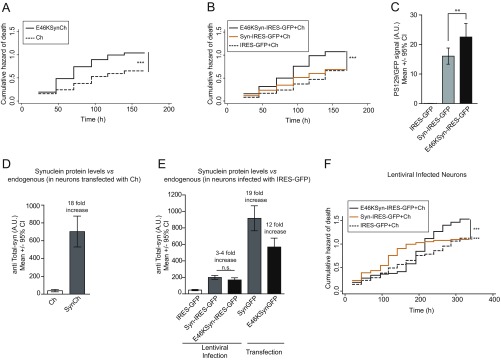
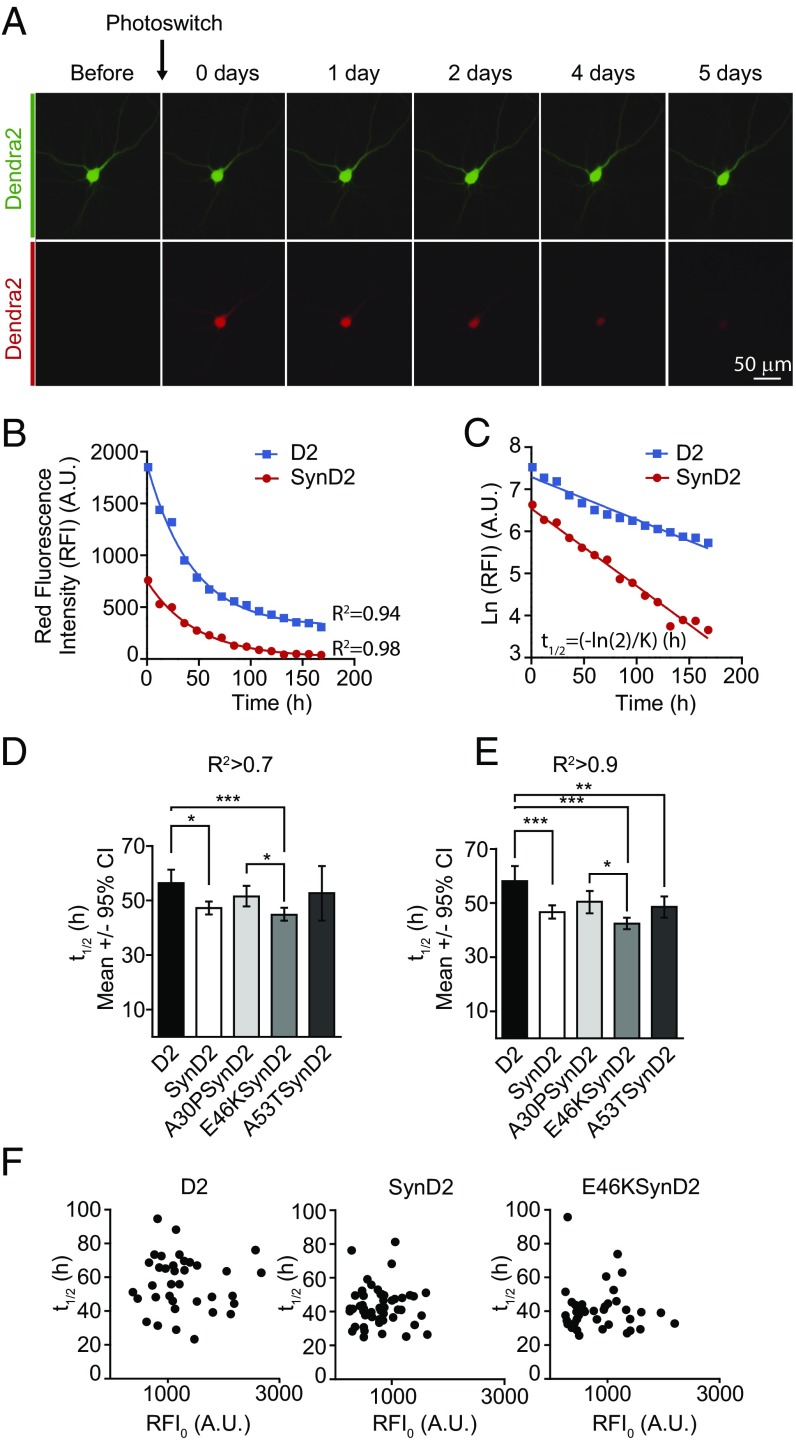
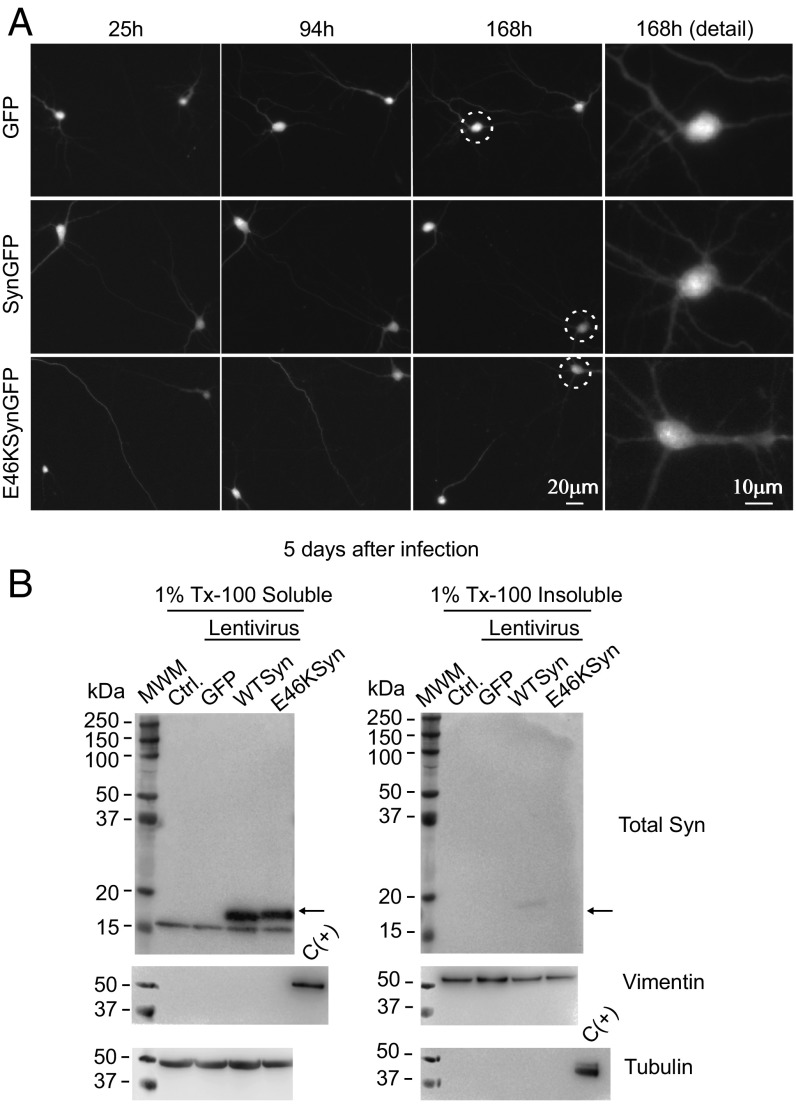
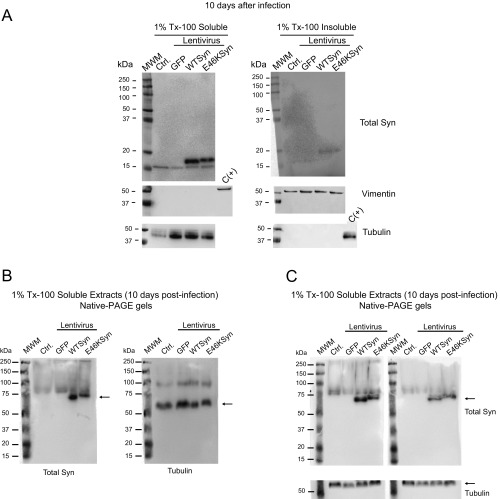
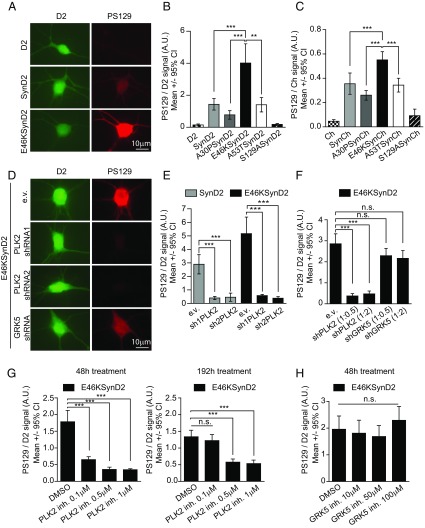
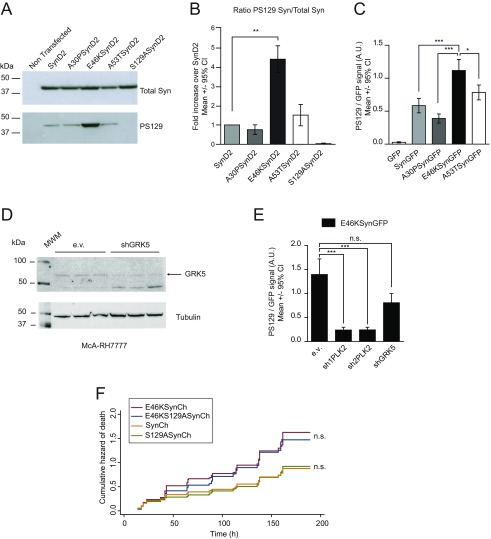
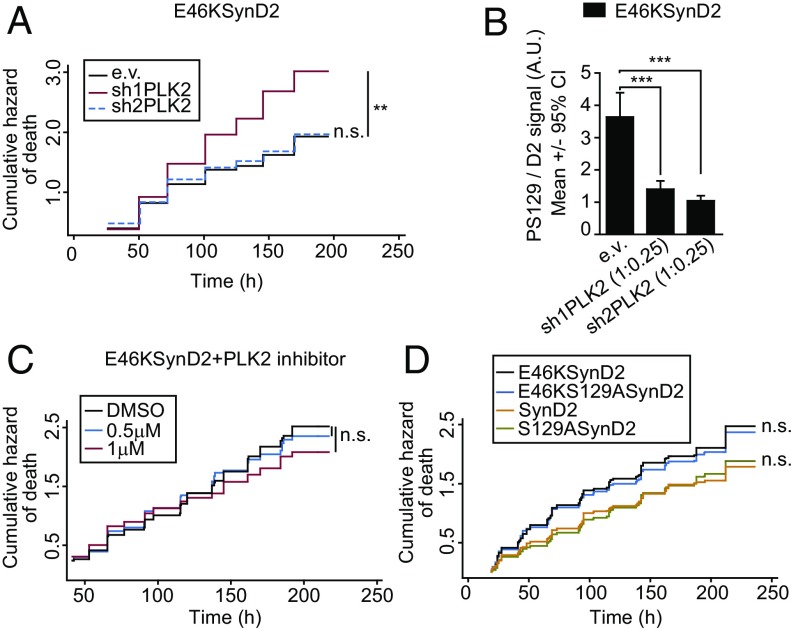
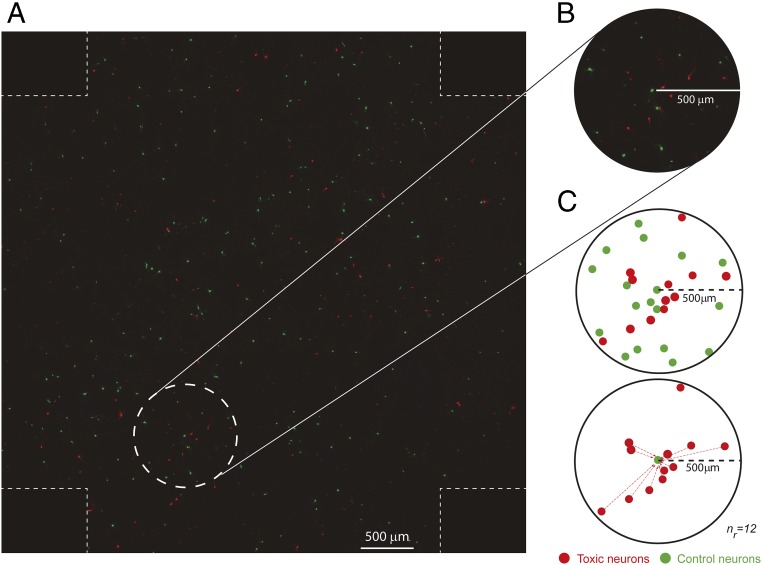
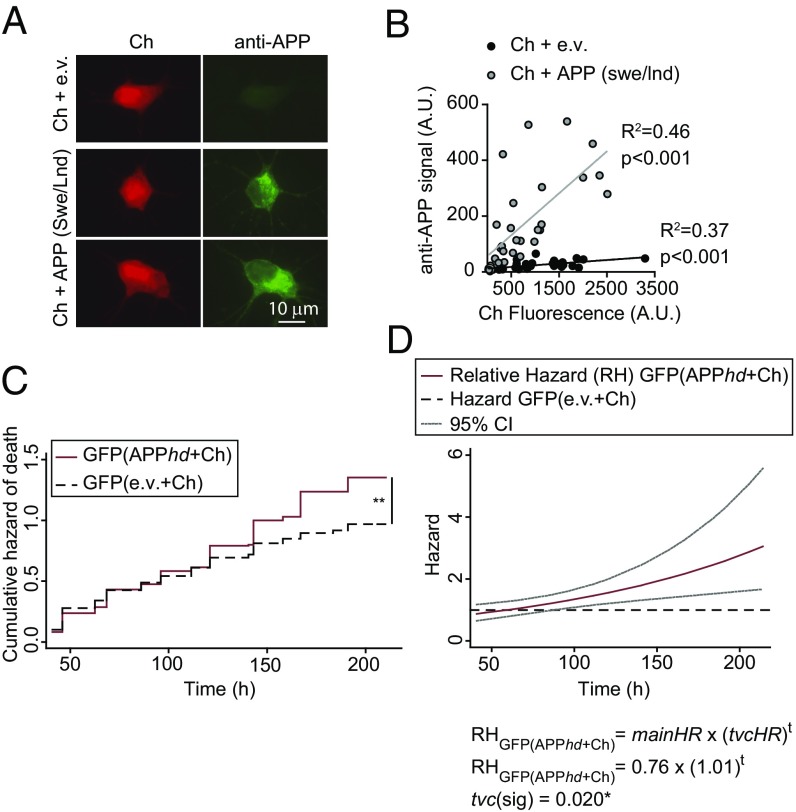
α-Synuclein (aSyn) is the main driver of neurodegenerative diseases known as "synucleinopathies," but the mechanisms underlying this toxicity remain poorly understood. To investigate aSyn toxic mechanisms, we have developed a primary neuronal model in which a longitudinal survival analysis can be performed by following the overexpression of fluorescently tagged WT or pathologically mutant aSyn constructs. Most aSyn mutations linked to neurodegenerative disease hindered neuronal survival in this model; of these mutations, the E46K mutation proved to be the most toxic. While E46K induced robust PLK2-dependent aSyn phosphorylation at serine 129, inhibiting this phosphorylation did not alleviate aSyn toxicity, strongly suggesting that this pathological hallmark of synucleinopathies is an epiphenomenon. Optical pulse-chase experiments with Dendra2-tagged aSyn versions indicated that the E46K mutation does not alter aSyn protein turnover. Moreover, since the mutation did not promote overt aSyn aggregation, we conclude that E46K toxicity was driven by soluble species. Finally, we developed an assay to assess whether neurons expressing E46K aSyn affect the survival of neighboring control neurons. Although we identified a minor non-cell-autonomous component spatially restricted to proximal neurons, most E46K aSyn toxicity was cell autonomous. Thus, we have been able to recapitulate the toxicity of soluble aSyn species at a stage preceding aggregation, detecting non-cell-autonomous toxicity and evaluating how some of the main aSyn hallmarks are related to neuronal survival.
Keywords: E46K mutation; alpha-synuclein; autonomous toxicity; neuronal death; serine 129 phosphorylation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity.Neurobiol Dis. 2020 Apr;137:104781. doi: 10.1016/j.nbd.2020.104781. Epub 2020 Jan 25. Neurobiol Dis. 2020. PMID: 31991248
-
Environmental and genetic factors support the dissociation between α-synuclein aggregation and toxicity.Proc Natl Acad Sci U S A. 2016 Oct 18;113(42):E6506-E6515. doi: 10.1073/pnas.1606791113. Epub 2016 Oct 5. Proc Natl Acad Sci U S A. 2016. PMID: 27708160 Free PMC article.
-
Stabilization of 14-3-3 protein-protein interactions with Fusicoccin-A decreases alpha-synuclein dependent cell-autonomous death in neuronal and mouse models.Neurobiol Dis. 2023 Jul;183:106166. doi: 10.1016/j.nbd.2023.106166. Epub 2023 May 26. Neurobiol Dis. 2023. PMID: 37245833
-
Lewy body-associated proteins: victims, instigators, or innocent bystanders? The case of AIMP2 and alpha-synuclein.Neurobiol Dis. 2021 Aug;156:105417. doi: 10.1016/j.nbd.2021.105417. Epub 2021 Jun 6. Neurobiol Dis. 2021. PMID: 34102275 Review.
-
Traffic jams and the complex role of α-Synuclein aggregation in Parkinson disease.Small GTPases. 2017 Apr 3;8(2):78-84. doi: 10.1080/21541248.2016.1199191. Epub 2016 Jun 17. Small GTPases. 2017. PMID: 27314512 Free PMC article. Review.
Cited by
-
Parkinson's disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure.Nat Commun. 2020 May 26;11(1):2643. doi: 10.1038/s41467-020-16386-3. Nat Commun. 2020. PMID: 32457390 Free PMC article.
-
E46K α-Synuclein Mutation Fails to Promote Neurite Outgrowth by Not Inducing Cdc42EP2 Expression, Unlike Wild-Type or A53T α-Synuclein in SK-N-SH Cells.Brain Sci. 2024 Dec 25;15(1):9. doi: 10.3390/brainsci15010009. Brain Sci. 2024. PMID: 39851377 Free PMC article.
-
Dynamic behaviors of α-synuclein and tau in the cellular context: New mechanistic insights and therapeutic opportunities in neurodegeneration.Neurobiol Dis. 2019 Dec;132:104543. doi: 10.1016/j.nbd.2019.104543. Epub 2019 Jul 24. Neurobiol Dis. 2019. PMID: 31351173 Free PMC article. Review.
-
Rationally Designed Variants of α-Synuclein Illuminate Its in vivo Structural Properties in Health and Disease.Front Neurosci. 2018 Sep 25;12:623. doi: 10.3389/fnins.2018.00623. eCollection 2018. Front Neurosci. 2018. PMID: 30319334 Free PMC article. Review.
-
Skin alpha-synuclein deposit patterns: A predictor of Parkinson's disease subtypes.EBioMedicine. 2022 Jun;80:104076. doi: 10.1016/j.ebiom.2022.104076. Epub 2022 May 26. EBioMedicine. 2022. PMID: 35644126 Free PMC article. Review.
References
-
- Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011;122:187–204. - PubMed
-
- Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. - PubMed
-
- Nalls MA, et al. International Parkinson’s Disease Genomics Consortium (IPDGC); Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI); 23andMe; GenePD; NeuroGenetics Research Consortium (NGRC); Hussman Institute of Human Genomics (HIHG); Ashkenazi Jewish Dataset Investigator; Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE); North American Brain Expression Consortium (NABEC); United Kingdom Brain Expression Consortium (UKBEC); Greek Parkinson’s Disease Consortium; Alzheimer Genetic Analysis Group Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–993. - PMC - PubMed
-
- Maraganore DM, et al. Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
