

β‑thalassemia caused by compound heterozygous mutations and cured by bone marrow transplantation: A case report
- PMID: 28901454
- PMCID: PMC5865824
- DOI: 10.3892/mmr.2017.7476
β‑thalassemia caused by compound heterozygous mutations and cured by bone marrow transplantation: A case report
Abstract
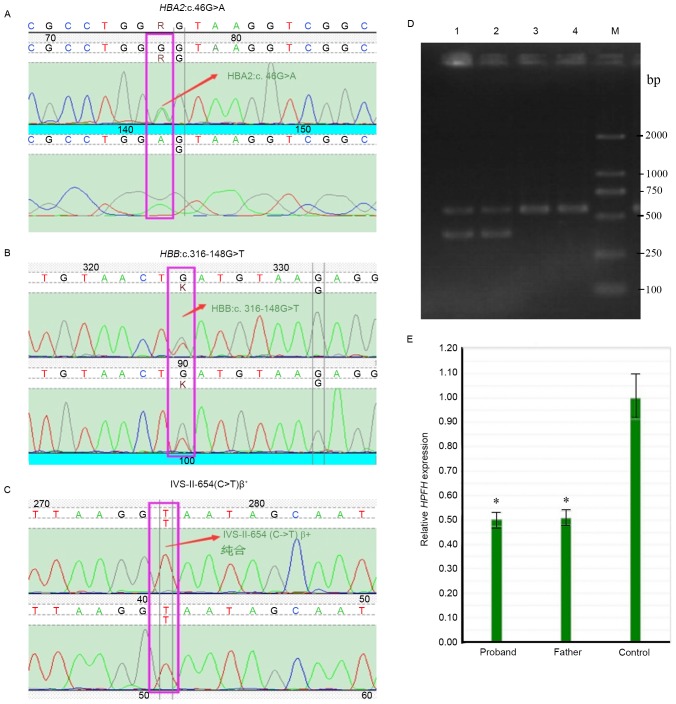
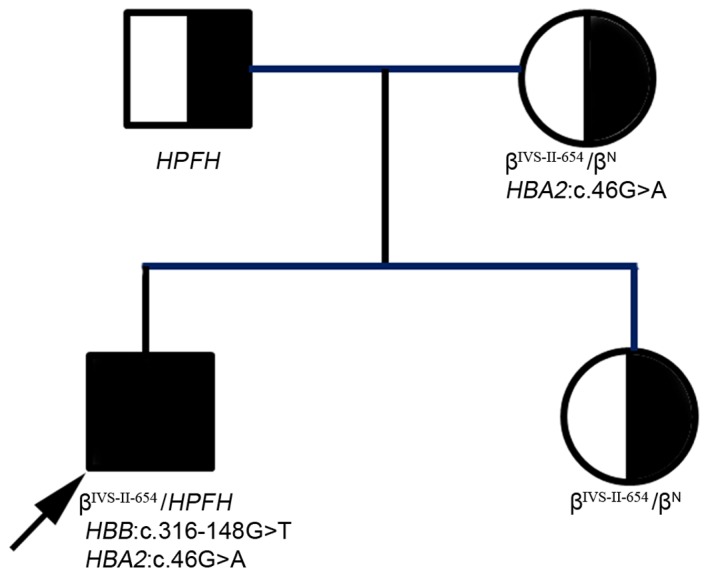
In the present study, a rare familial case of severe thalassemia with compound spontaneous mutations is reported. A 2.5‑year‑old boy, who suffered from severe anemia with yellowish skin, enlarged liver and spleen, was provided with a blood transfusion every 20 days to maintain hemoglobin levels between 90 and 100 g/l. Sanger sequencing combined with reverse transcription‑quantitative polymerase chain reaction (RT‑qPCR) and Gap‑PCR revealed that the proband was a carrier of 4 compound heterozygous mutations: Hemoglobin subunit β (HBB):IVS‑II‑654(C>T)β+; Southeast Asian‑type‑hereditary persistence of fetal hemoglobin (SEA‑HPFH); HBB:c316‑148G>T; hemoglobin subunit α2 (HBA2):c.46G>A. The father of the proband was identified as a carrier of the heterozygous SEA‑HPFH mutation, the mother was a carrier of compound heterozygous mutations of HBB:IVS‑II‑654(C>T) and HBA2:c.46G>A, and the elder sister was heterozygous for HBB:IVS‑II‑654(C>T)β+. Based on these genetic results, it was determined that the proband had both of heavy β‑thalassemia and α‑thalassemia. Upon human leukocyte antigen matching, bone marrow transplantation (BMT) was successfully performed on the proband by selecting his HLA‑compatible sister as a donor. Following treatment, the proband was revealed to only carry the IVS‑II‑654(C>T)β+ heterozygous mutation, and further regular blood transfusions have been avoided; BMT results remained normal at six months follow‑up.
Figures

Similar articles
-
α‑thalassemia deletion [‑SEA (Southeast Asian)] and a compound heterozygote for the Chinese Gγ+(Aγδβ)0/βCD17‑thalassemia mutation: A case report.Mol Med Rep. 2023 Jun;27(6):112. doi: 10.3892/mmr.2023.12999. Epub 2023 Apr 21. Mol Med Rep. 2023. PMID: 37083078
-
Hb Midnapore [β53(D4)Ala→Val; HBB: c.161C>T]: A Novel Hemoglobin Variant with a Structural Abnormality Associated with IVS-I-5 (G>C) (HBB: c.92+5G>C) Found in a Bengali Indian Family.Hemoglobin. 2016 Sep;40(5):300-303. doi: 10.1080/03630269.2016.1243555. Hemoglobin. 2016. PMID: 27690257
-
Hereditary spherocytosis overlooked for 7 years in a pediatric patient with β-thalassemia trait and novel compound heterozygous mutations of SPTA1 gene.Hematology. 2020 Dec;25(1):438-445. doi: 10.1080/16078454.2020.1846874. Hematology. 2020. PMID: 33210974
-
Ten Years of Routine α- and β-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations.Hemoglobin. 2016;40(2):75-84. doi: 10.3109/03630269.2015.1113990. Epub 2015 Dec 4. Hemoglobin. 2016. PMID: 26635043 Review.
-
[A family with two children diagnosed with aspartylglucosaminuria-case report and literature review].Zhonghua Er Ke Za Zhi. 2014 Jun;52(6):455-9. Zhonghua Er Ke Za Zhi. 2014. PMID: 25190167 Review. Chinese.
References
-
- Muncie HL, Jr, Campbell J. Alpha and beta thalassemia. Am Fam Physician. 2009;80:339–344. - PubMed
-
- Cai R, Li L, Liang X, Liu Z, Su L, Li W, Zhu Q, Mo Q, Pan L, Ouyang H, et al. Prevalence survey and molecular characterization of alpha and beta thalassemia in Liuzhou city of Guangxi. Zhonghua Liu Xing Bing Xue Za Zhi. 2002;23:281–285. (In Chinese) - PubMed
-
- Xu XM, Zhou YQ, Luo GX, Liao C, Zhou M, Chen PY, Lu JP, Jia SQ, Xiao GF, Shen X, et al. The prevalence and spectrum of alpha and beta thalassaemia in Guangdong Province: Implications for the future health burden and population screening. J Clin Pathol. 2004;57:517–522. doi: 10.1136/jcp.2003.014456. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
