

Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity
- PMID: 28904012
- PMCID: PMC5630028
- DOI: 10.1101/gr.220533.117
Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity
Abstract
The tumor suppressor TP53 is the most frequently mutated gene product in human cancer. Close to half of all solid tumors carry inactivating mutations in the TP53 gene, while in the remaining cases, TP53 activity is abrogated by other oncogenic events, such as hyperactivation of its endogenous repressors MDM2 or MDM4. Despite identification of hundreds of genes regulated by this transcription factor, it remains unclear which direct target genes and downstream pathways are essential for the tumor suppressive function of TP53. We set out to address this problem by generating multiple genomic data sets for three different cancer cell lines, allowing the identification of distinct sets of TP53-regulated genes, from early transcriptional targets through to late targets controlled at the translational level. We found that although TP53 elicits vastly divergent signaling cascades across cell lines, it directly activates a core transcriptional program of ∼100 genes with diverse biological functions, regardless of cell type or cellular response to TP53 activation. This core program is associated with high-occupancy TP53 enhancers, high levels of paused RNA polymerases, and accessible chromatin. Interestingly, two different shRNA screens failed to identify a single TP53 target gene required for the anti-proliferative effects of TP53 during pharmacological activation in vitro. Furthermore, bioinformatics analysis of thousands of cancer genomes revealed that none of these core target genes are frequently inactivated in tumors expressing wild-type TP53. These results support the hypothesis that TP53 activates a genetically robust transcriptional program with highly distributed tumor suppressive functions acting in diverse cellular contexts.
© 2017 Andrysik et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
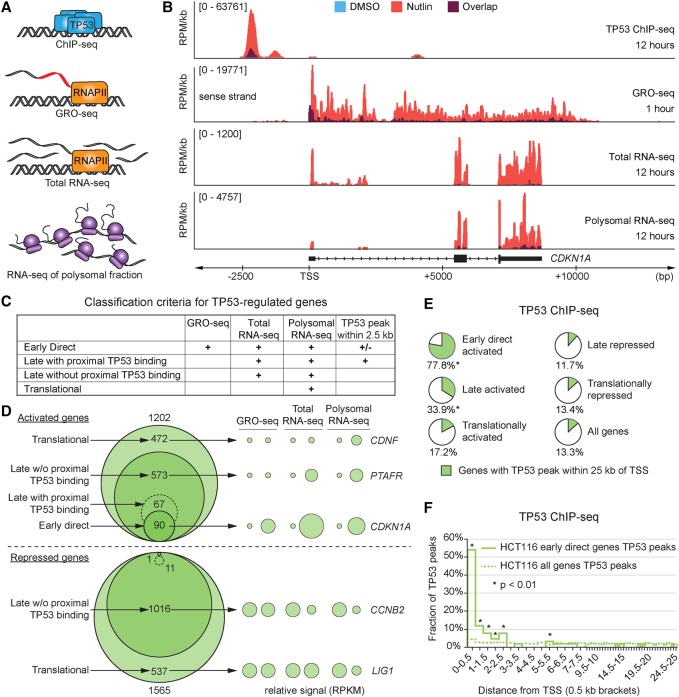
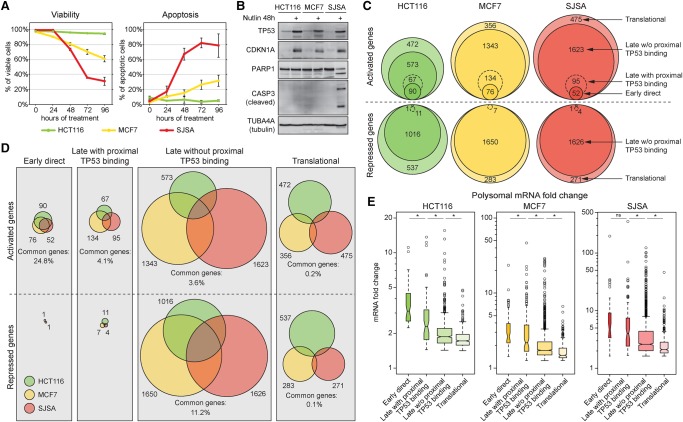
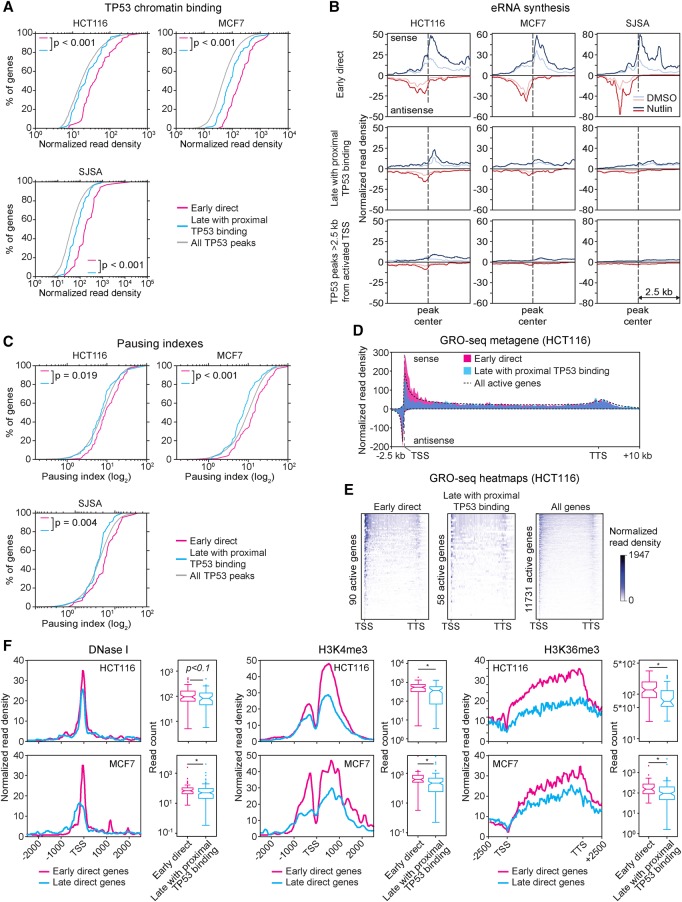
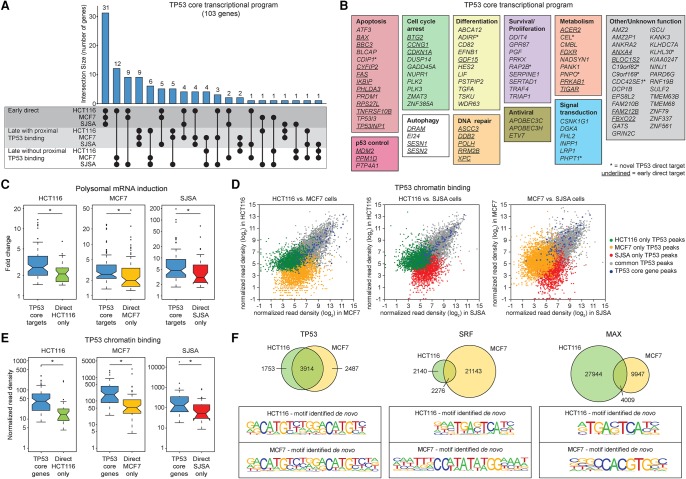
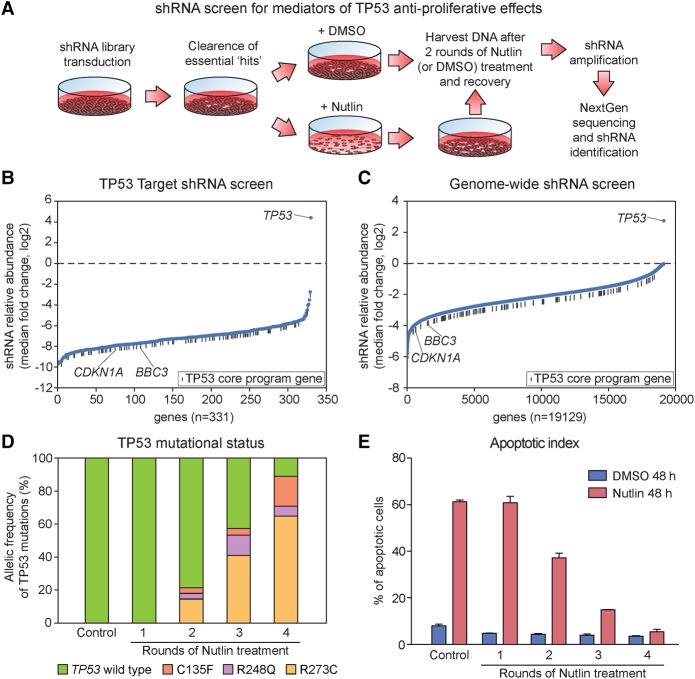
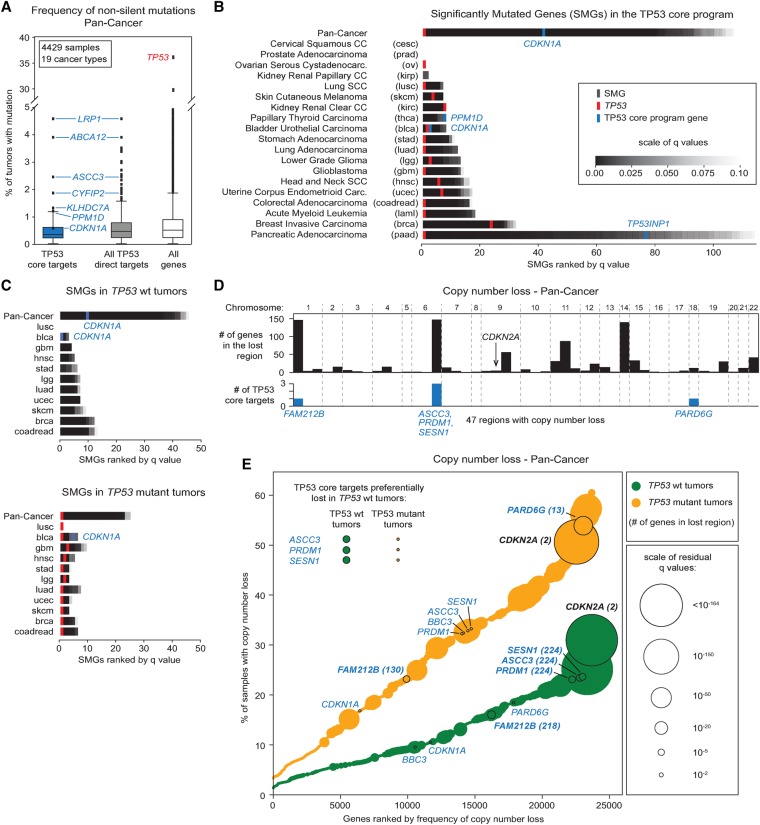
References
-
- Brummelkamp TR, Fabius AW, Mullenders J, Madiredjo M, Velds A, Kerkhoven RM, Bernards R, Beijersbergen RL. 2006. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol 2: 202–206. - PubMed
-
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous