

Parallel Evolution of Group B Streptococcus Hypervirulent Clonal Complex 17 Unveils New Pathoadaptive Mutations
- PMID: 28904998
- PMCID: PMC5585690
- DOI: 10.1128/mSystems.00074-17
Parallel Evolution of Group B Streptococcus Hypervirulent Clonal Complex 17 Unveils New Pathoadaptive Mutations
Abstract
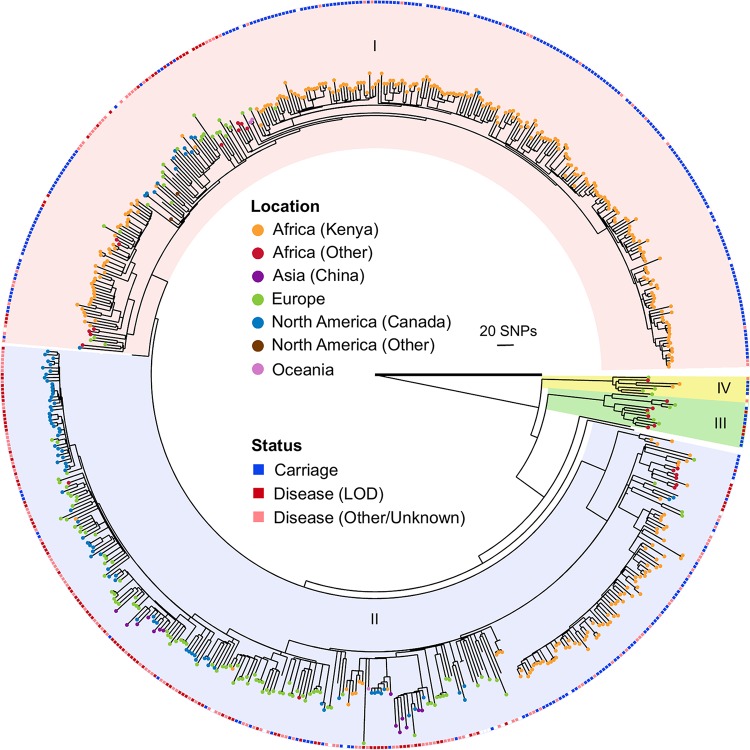
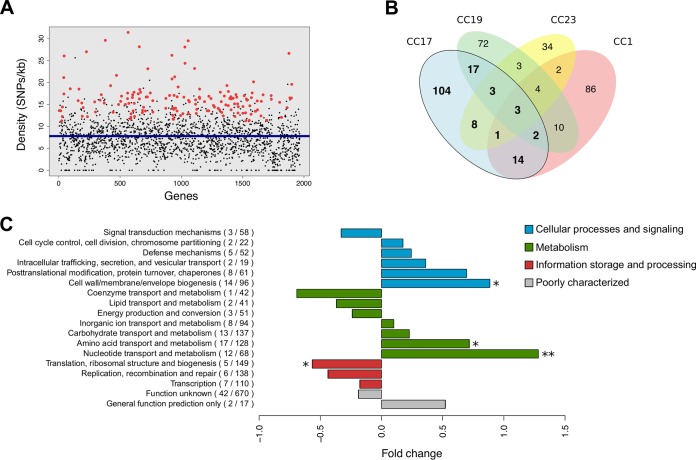
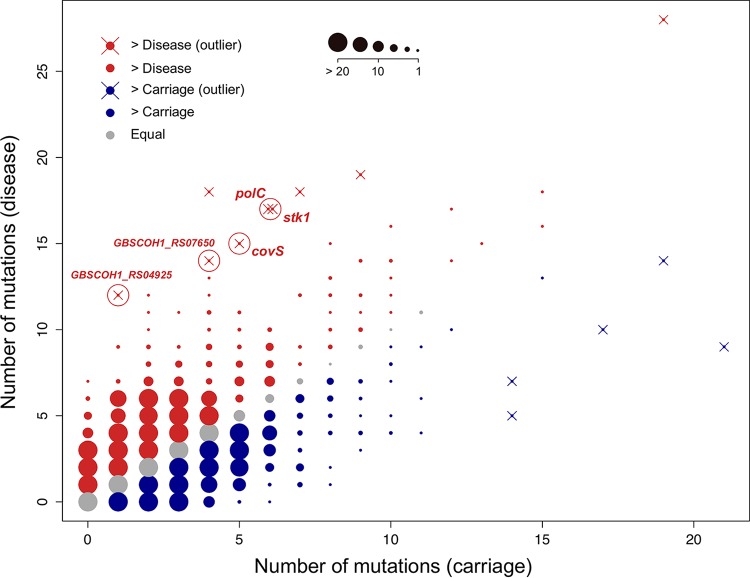
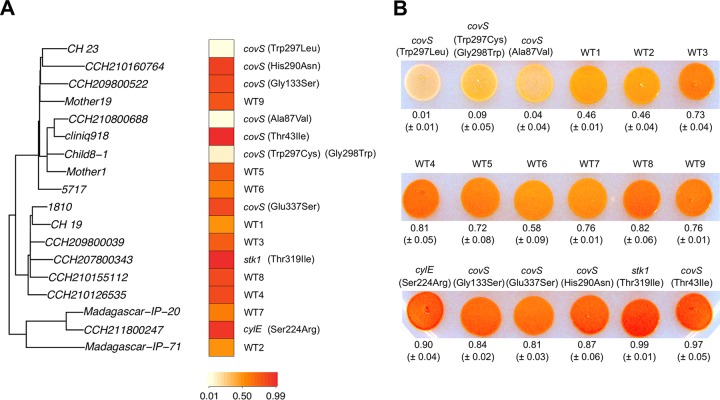
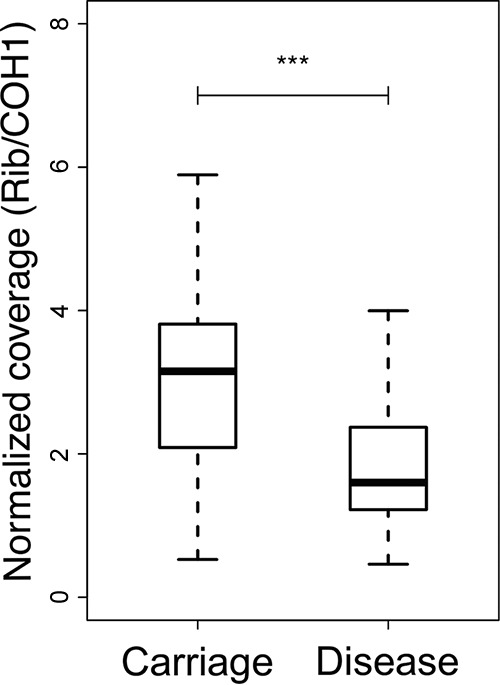
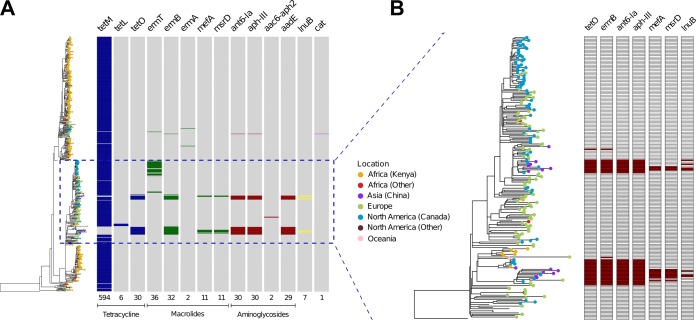
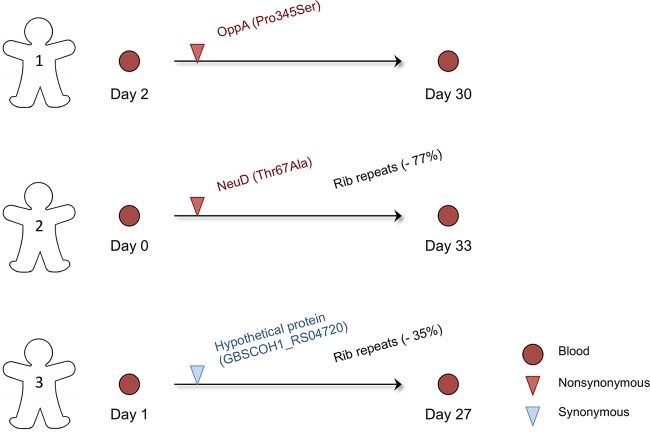
Group B Streptococcus (GBS) is a commensal of the gastrointestinal and genitourinary tracts, while a prevailing cause of neonatal disease worldwide. Of the various clonal complexes (CCs), CC17 is overrepresented in GBS-infected newborns for reasons that are still largely unknown. Here, we report a comprehensive genomic analysis of 626 CC17 isolates collected worldwide, identifying the genetic traits behind their successful adaptation to humans and the underlying differences between carriage and clinical strains. Comparative analysis with 923 GBS genomes belonging to CC1, CC19, and CC23 revealed that the evolution of CC17 is distinct from that of other human-adapted lineages and recurrently targets functions related to nucleotide and amino acid metabolism, cell adhesion, regulation, and immune evasion. We show that the most distinctive features of disease-specific CC17 isolates were frequent mutations in the virulence-associated CovS and Stk1 kinases, underscoring the crucial role of the entire CovRS regulatory pathway in modulating the pathogenicity of GBS. Importantly, parallel and convergent evolution of major components of the bacterial cell envelope, such as the capsule biosynthesis operon, the pilus, and Rib, reflects adaptation to host immune pressures and should be taken into account in the ongoing development of a GBS vaccine. The presence of recurrent targets of evolution not previously implicated in virulence also opens the way for uncovering new functions involved in host colonization and GBS pathogenesis. IMPORTANCE The incidence of group B Streptococcus (GBS) neonatal disease continues to be a significant cause of concern worldwide. Strains belonging to clonal complex 17 (CC17) are the most frequently responsible for GBS infections in neonates, especially among late-onset disease cases. Therefore, we undertook the largest genomic study of GBS CC17 strains to date to decipher the genetic bases of their remarkable colonization and infection ability. We show that crucial functions involved in different steps of the colonization or infection process of GBS are distinctly mutated during the adaptation of CC17 to the human host. In particular, our results implicate the CovRS two-component regulator of virulence in the differentiation between carriage- and disease-associated isolates. Not only does this work raise important implications for the ongoing development of a vaccine against GBS but might also drive the discovery of key functions for GBS adaptation and pathogenesis that have been overlooked until now. Author Video: An author video summary of this article is available.
Keywords: CovR; GBS vaccine; ST17; antibiotic resistance; eubacteria; evolution; genomics; group B Streptococcus; virulence.
Figures
References
-
- Edwards MS, Baker CJ. 2005. Group B streptococcal infections, p 1091–1156. In Klein JO, Remington JS (ed), Infectious diseases of the fetus and newborn infant. W. B. Saunders, ; Philadelphia, PA.
-
- Kobayashi M, Schrag SJ, Alderson MR, Madhi SA, Baker CJ, Sobanjo-ter Meulen A, Kaslow DC, Smith PG, Moorthy VS, Vekemans J 22 December 2016. WHO consultation on group B Streptococcus vaccine development: report from a meeting held on 27–28 April 2016. Vaccine. doi:10.1016/j.vaccine.2016.12.029. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
