

Coarse-grained molecular dynamics studies of the structure and stability of peptide-based drug amphiphile filaments
- PMID: 28905963
- PMCID: PMC5665727
- DOI: 10.1039/c7sm00943g
Coarse-grained molecular dynamics studies of the structure and stability of peptide-based drug amphiphile filaments
Abstract
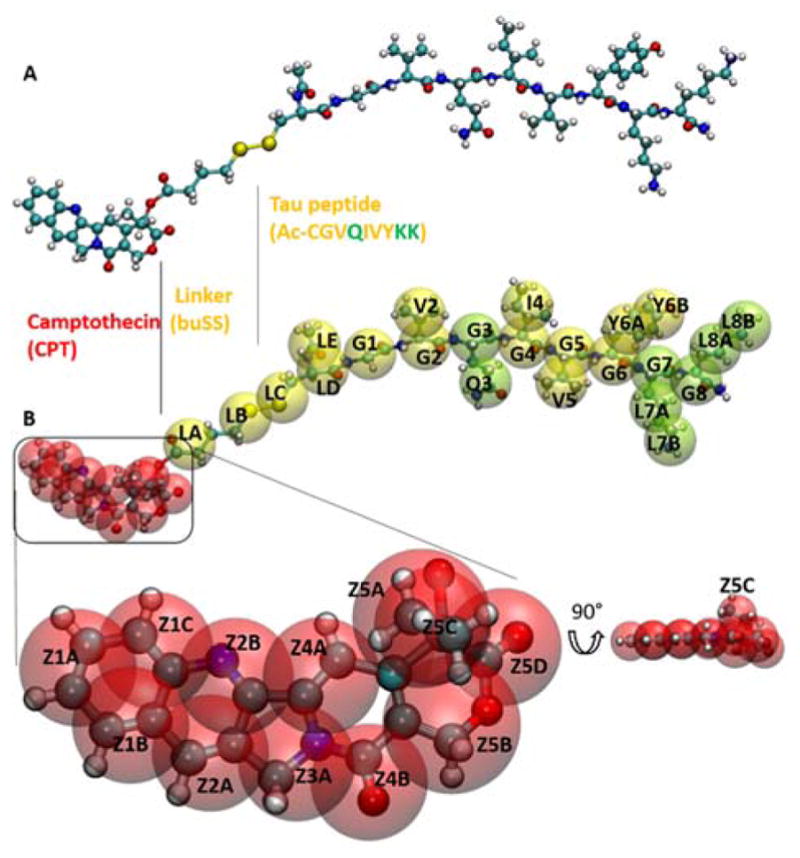
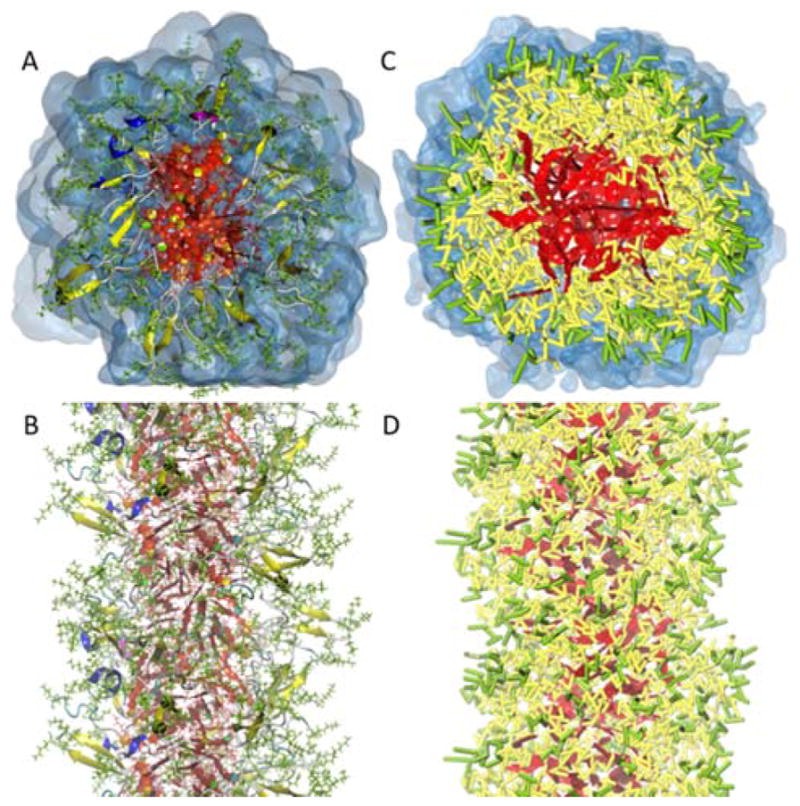
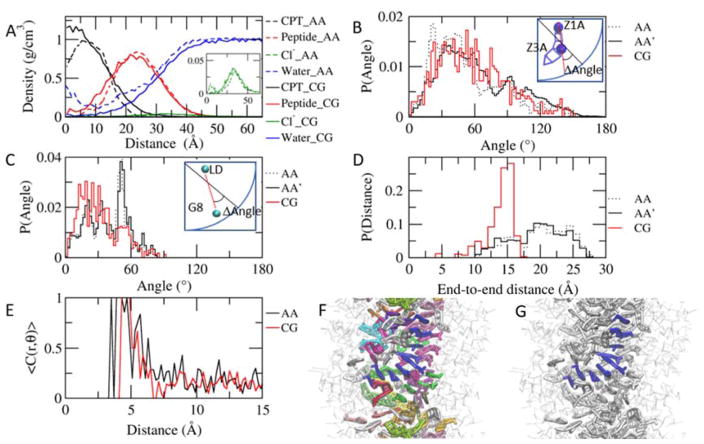
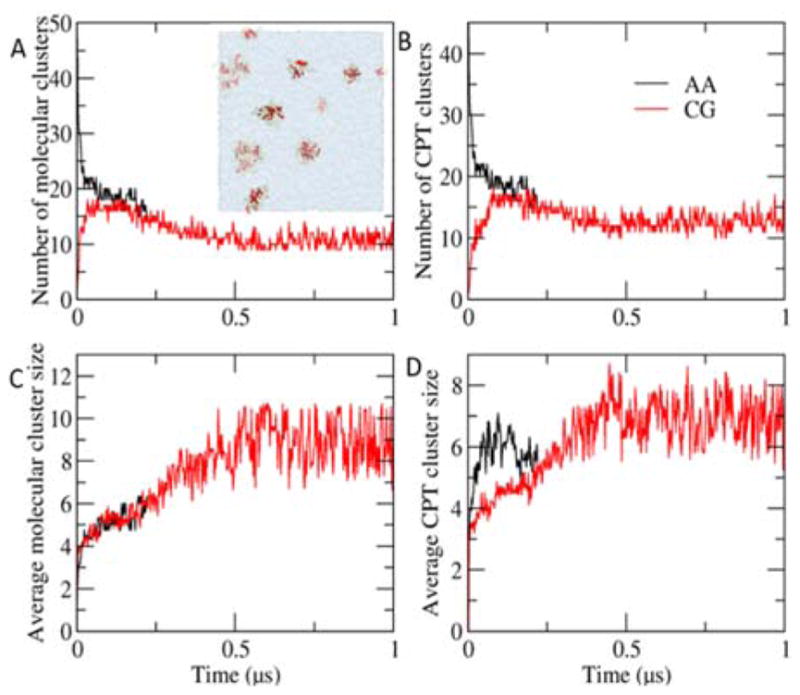
Peptide-based supramolecular filaments, in particular filaments self-assembled by drug amphiphiles (DAs), possess great potential in the field of drug delivery. These filaments possess one hundred percent drug loading, with a release mechanism that can be tuned based on the dissociation of the supramolecular filaments and the degradation of the DAs [Cheetham et al., J. Am. Chem. Soc., 2013, 135(8), 2907]. Recently, much attention has been drawn to the competing intermolecular interactions that drive the self-assembly of peptide-based amphiphiles into supramolecular filaments. Recently, we reported on long-time atomistic molecular dynamics simulations to characterize the structure and growth of chiral filaments by the self-assembly of a DA containing the aromatic anti-cancer drug camptothecin [Kang et al., Macromolecules, 2016, 49(3), 994]. We found that the π-π stacking of the aromatic drug governs the early stages of the self-assembly process, while also contributing towards the chirality of the self-assembled filament. Based on these all-atomistic simulations, we now build a chemically accurate coarse-grained model that can capture the structure and stability of these supramolecular filaments at long time-scales (microseconds). These coarse-grained models successfully recapitulate the growth of the molecular clusters (and their elongation trends) compared with previously reported atomistic simulations. Furthermore, the interfacial structure and the helicity of the filaments are conserved. Next, we focus on characterization of the disassembly process of a 0.675 μm DA filament at microsecond time-scales. These results provide very useful tools for the rational design of functional supramolecular filaments, in particular supramolecular filaments for drug delivery applications.
Figures
Similar articles
-
Molecular simulations of peptide amphiphiles.Org Biomol Chem. 2017 Oct 4;15(38):7993-8005. doi: 10.1039/c7ob01290j. Org Biomol Chem. 2017. PMID: 28853474 Free PMC article. Review.
-
Rational Coarse-Grained Molecular Dynamics Simulations of Supramolecular Anticancer Nanotubes.J Phys Chem B. 2019 Dec 19;123(50):10582-10593. doi: 10.1021/acs.jpcb.9b07417. Epub 2019 Dec 10. J Phys Chem B. 2019. PMID: 31749360
-
Molecular Dynamics Simulations of Supramolecular Anticancer Nanotubes.J Chem Inf Model. 2018 Jun 25;58(6):1164-1168. doi: 10.1021/acs.jcim.8b00193. Epub 2018 Jun 6. J Chem Inf Model. 2018. PMID: 29856610 Free PMC article.
-
Modeling Interactions within and between Peptide Amphiphile Supramolecular Filaments.J Phys Chem B. 2022 Jan 27;126(3):650-659. doi: 10.1021/acs.jpcb.1c09258. Epub 2022 Jan 14. J Phys Chem B. 2022. PMID: 35029997
-
Peptide Amphiphiles for Pharmaceutical Applications.Curr Med Chem. 2024;31(11):1332-1347. doi: 10.2174/0929867330666230408203820. Curr Med Chem. 2024. PMID: 37031390 Review.
Cited by
-
Peptide Self-Assembled Nanocarriers for Cancer Drug Delivery.J Phys Chem B. 2023 Mar 9;127(9):1857-1871. doi: 10.1021/acs.jpcb.2c06751. Epub 2023 Feb 22. J Phys Chem B. 2023. PMID: 36812392 Free PMC article. Review.
-
Mechanisms of Scaffold-Mediated Microcompartment Assembly and Size Control.ACS Nano. 2021 Mar 23;15(3):4197-4212. doi: 10.1021/acsnano.0c05715. Epub 2021 Mar 8. ACS Nano. 2021. PMID: 33683101 Free PMC article.
-
Energy Landscapes of Supramolecular Peptide-Drug Conjugates Directed by Linker Selection and Drug Topology.ACS Nano. 2022 Jun 28;16(6):9546-9558. doi: 10.1021/acsnano.2c02804. Epub 2022 May 31. ACS Nano. 2022. PMID: 35639629 Free PMC article.
-
Molecular simulations of peptide amphiphiles.Org Biomol Chem. 2017 Oct 4;15(38):7993-8005. doi: 10.1039/c7ob01290j. Org Biomol Chem. 2017. PMID: 28853474 Free PMC article. Review.
-
Modelling of interactions between Aβ(25-35) peptide and phospholipid bilayers: effects of cholesterol and lipid saturation.RSC Adv. 2020 Jan 23;10(7):3902-3915. doi: 10.1039/c9ra06424a. eCollection 2020 Jan 22. RSC Adv. 2020. PMID: 35492630 Free PMC article.
References
-
- Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. - PubMed
-
- Trent A, Marullo R, Lin B, Black M, Tirrell M. Soft Matter. 2011;7:9572–9572.
-
- Kokkoli E, Mardilovich A, Wedekind A, Rexeisen EL, Garg A, Craig JA. Soft Matter. 2006;2:1015–1024. - PubMed
-
- Gao C, Li H, Li Y, Kewalramani S, Palmer LC, Dravid VP, Stupp SI, Olvera de la Cruz M, Bedzyk MJ. J Phys Chem B. 2017;121:1623–1628. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous