

Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift
- PMID: 28910616
- PMCID: PMC5678753
- DOI: 10.1016/j.neuron.2017.07.010
Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift
Abstract
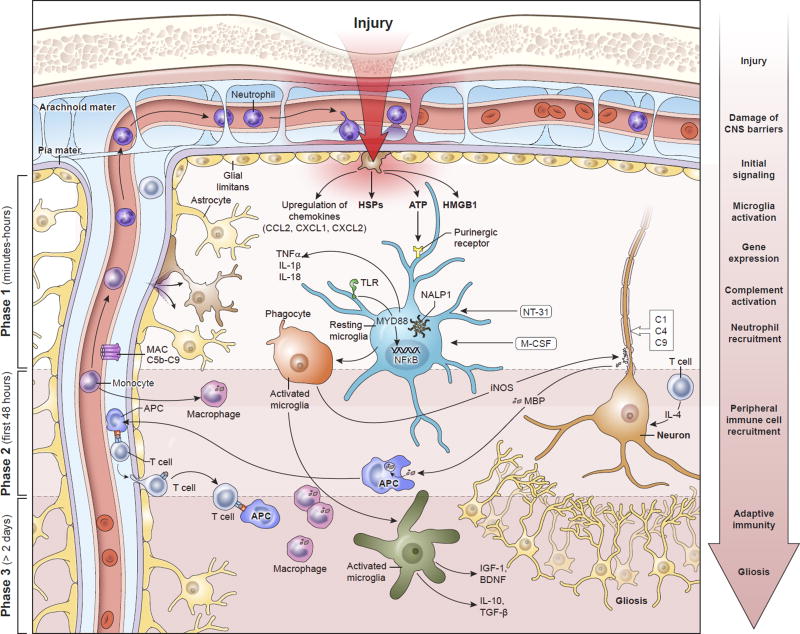
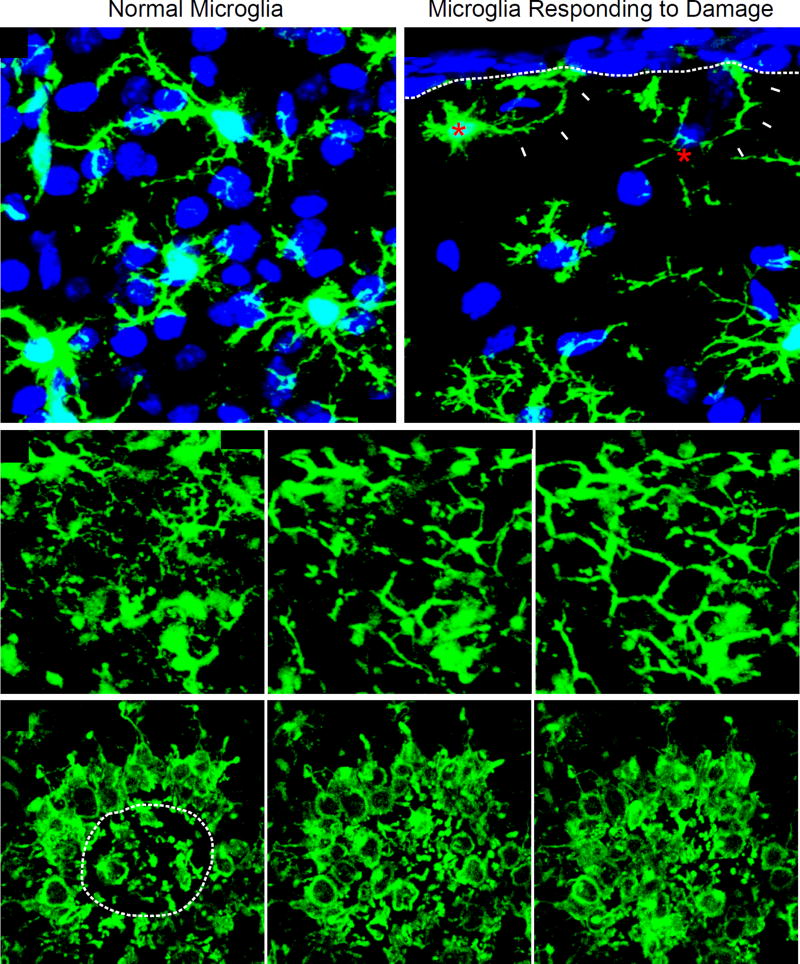
Traumatic brain injury (TBI) is a leading cause of morbidity and disability, with a considerable socioeconomic burden. Heterogeneity of pathoanatomical subtypes and diversity in the pathogenesis and extent of injury contribute to differences in the course and outcome of TBI. Following the primary injury, extensive and lasting damage is sustained through a complex cascade of events referred to as "secondary injury." Neuroinflammation is proposed as an important manipulable aspect of secondary injury in animal and human studies. Because neuroinflammation can be detrimental or beneficial, before developing immunomodulatory therapies, it is necessary to better understand the timing and complexity of the immune responses that follow TBI. With a rapidly increasing body of literature, there is a need for a clear summary of TBI neuroimmunology. This review presents our current understanding of the immune response to TBI in a chronological and compartment-based manner, highlighting early changes in gene expression and initial signaling pathways that lead to activation of innate and adaptive immunity. Based on recent advances in our understanding of innate immune cell activation, we propose a new paradigm to study innate immune cells following TBI that moves away from the existing M1/M2 classification of activation states toward a stimulus- and disease-specific understanding of polarization state based on transcriptomic and proteomic profiling.
Keywords: M1; M2; TBI; astrocytes; microglia; models; neurodegeneration; neuroimmunology; neuroinflammation; neutrophils; transcriptome.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Ambrosini E, Aloisi F. Chemokines and glial cells: a complex network in the central nervous system. Neurochem Res. 2004;29:1017–1038. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
