

Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution
- PMID: 28911207
- PMCID: PMC5755684
- DOI: 10.1093/hmg/ddx280
Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution
Abstract
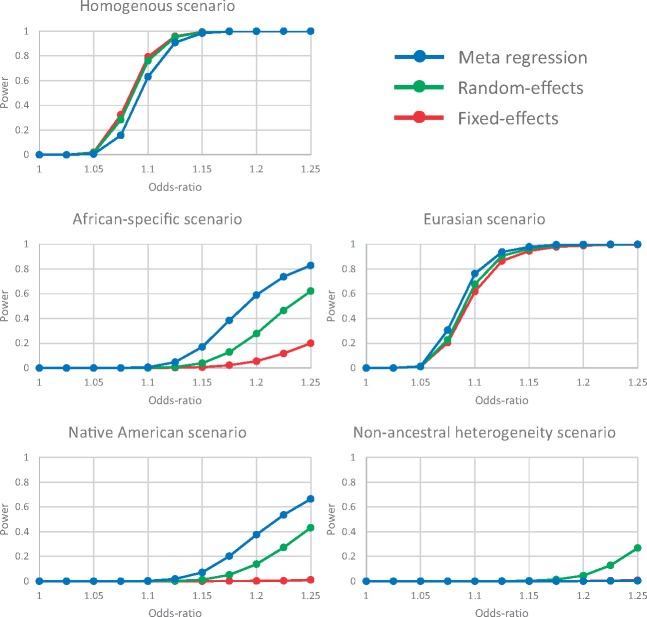
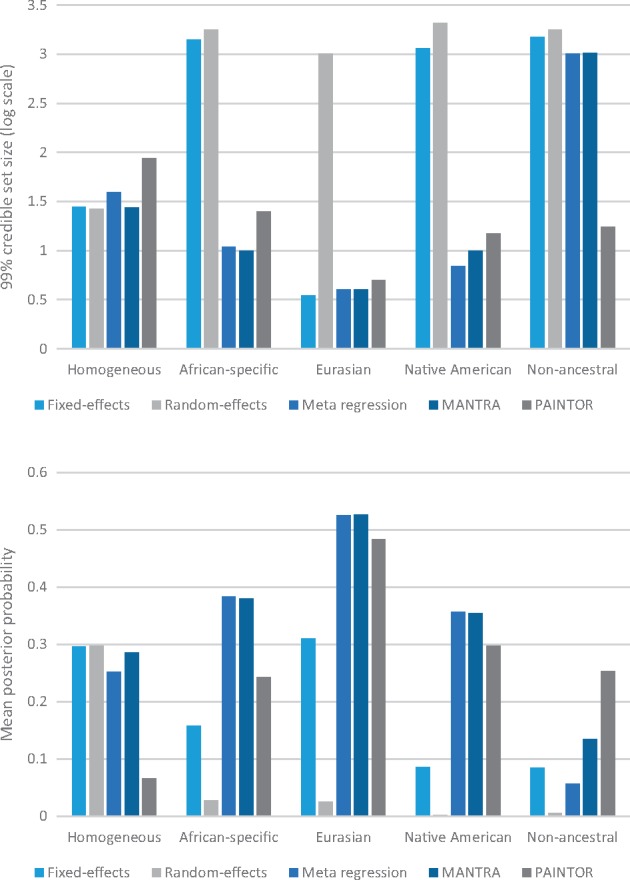
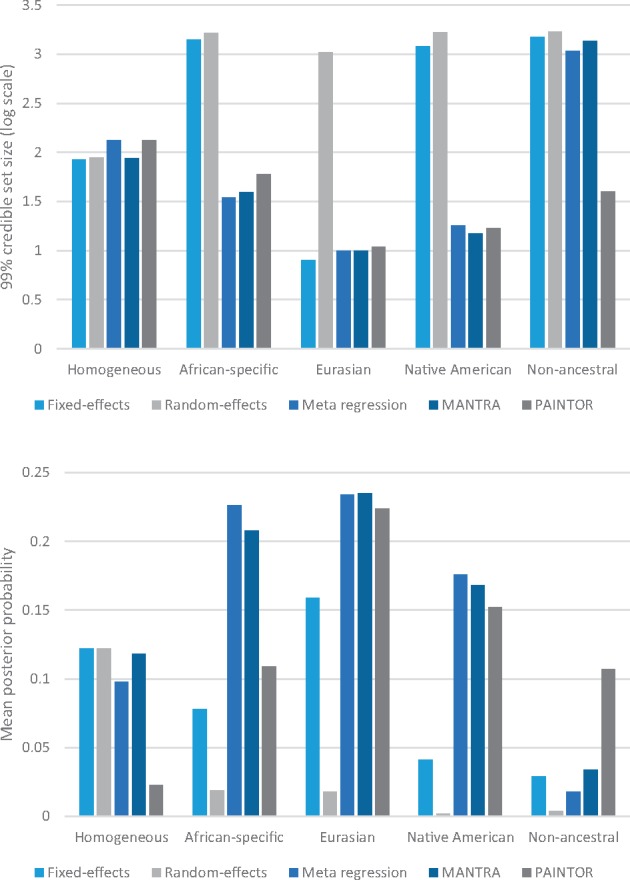
Trans-ethnic meta-analysis of genome-wide association studies (GWAS) across diverse populations can increase power to detect complex trait loci when the underlying causal variants are shared between ancestry groups. However, heterogeneity in allelic effects between GWAS at these loci can occur that is correlated with ancestry. Here, a novel approach is presented to detect SNP association and quantify the extent of heterogeneity in allelic effects that is correlated with ancestry. We employ trans-ethnic meta-regression to model allelic effects as a function of axes of genetic variation, derived from a matrix of mean pairwise allele frequency differences between GWAS, and implemented in the MR-MEGA software. Through detailed simulations, we demonstrate increased power to detect association for MR-MEGA over fixed- and random-effects meta-analysis across a range of scenarios of heterogeneity in allelic effects between ethnic groups. We also demonstrate improved fine-mapping resolution, in loci containing a single causal variant, compared to these meta-analysis approaches and PAINTOR, and equivalent performance to MANTRA at reduced computational cost. Application of MR-MEGA to trans-ethnic GWAS of kidney function in 71,461 individuals indicates stronger signals of association than fixed-effects meta-analysis when heterogeneity in allelic effects is correlated with ancestry. Application of MR-MEGA to fine-mapping four type 2 diabetes susceptibility loci in 22,086 cases and 42,539 controls highlights: (i) strong evidence for heterogeneity in allelic effects that is correlated with ancestry only at the index SNP for the association signal at the CDKAL1 locus; and (ii) 99% credible sets with six or fewer variants for five distinct association signals.
© The Author 2017. Published by Oxford University Press.
Figures
References
-
- Ntzani E.E., Liberopoulos G., Manolio T.A., Ioannidis J.P.A. (2012) Consistency of genome-wide associations across major ancestral groups. Hum. Genet., 131, 1057–1071. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
