

Pediatric Cardiomyopathies
- PMID: 28912187
- PMCID: PMC5657298
- DOI: 10.1161/CIRCRESAHA.116.309386
Pediatric Cardiomyopathies
Abstract
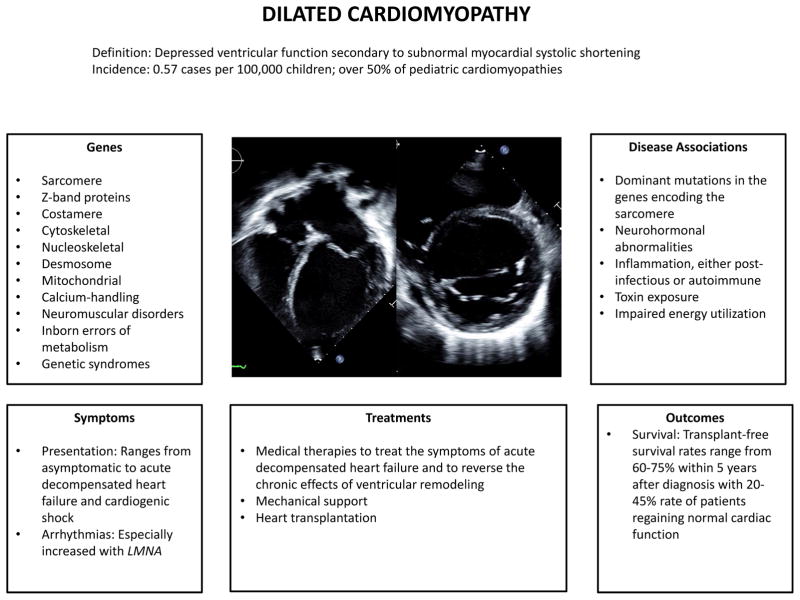
Pediatric cardiomyopathies are rare diseases with an annual incidence of 1.1 to 1.5 per 100 000. Dilated and hypertrophic cardiomyopathies are the most common; restrictive, noncompaction, and mixed cardiomyopathies occur infrequently; and arrhythmogenic right ventricular cardiomyopathy is rare. Pediatric cardiomyopathies can result from coronary artery abnormalities, tachyarrhythmias, exposure to infection or toxins, or secondary to other underlying disorders. Increasingly, the importance of genetic mutations in the pathogenesis of isolated or syndromic pediatric cardiomyopathies is becoming apparent. Pediatric cardiomyopathies often occur in the absence of comorbidities, such as atherosclerosis, hypertension, renal dysfunction, and diabetes mellitus; as a result, they offer insights into the primary pathogenesis of myocardial dysfunction. Large international registries have characterized the epidemiology, cause, and outcomes of pediatric cardiomyopathies. Although adult and pediatric cardiomyopathies have similar morphological and clinical manifestations, their outcomes differ significantly. Within 2 years of presentation, normalization of function occurs in 20% of children with dilated cardiomyopathy, and 40% die or undergo transplantation. Infants with hypertrophic cardiomyopathy have a 2-year mortality of 30%, whereas death is rare in older children. Sudden death is rare. Molecular evidence indicates that gene expression differs between adult and pediatric cardiomyopathies, suggesting that treatment response may differ as well. Clinical trials to support evidence-based treatments and the development of disease-specific therapies for pediatric cardiomyopathies are in their infancy. This compendium summarizes current knowledge of the genetic and molecular origins, clinical course, and outcomes of the most common phenotypic presentations of pediatric cardiomyopathies and highlights key areas where additional research is required.
Clinical trial registration: URL: http://www.clinicaltrials.gov. Unique identifiers: NCT02549664 and NCT01912534.
Keywords: epidemiology; genetics; pediatrics.
© 2017 American Heart Association, Inc.
Figures




References
-
- Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD. The Incidence of Pediatric Cardiomyopathy in Two Regions of the United States. N Engl J Med. 2003;348:1647–1655. - PubMed
-
- Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG. The Epidemiology of Childhood Cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–1646. - PubMed
-
- Nugent AW, Daubeney PE, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG National Australian Childhood Cardiomyopathy S. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation. 2005;112:1332–8. - PubMed
-
- Daubeney PE, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG National Australian Childhood Cardiomyopathy S. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. 2006;114:2671–8. - PubMed
-
- Alexander PM, Daubeney PE, Nugent AW, Lee KJ, Turner C, Colan SD, Robertson T, Davis AM, Ramsay J, Justo R, Sholler GF, King I, Weintraub RG National Australian Childhood Cardiomyopathy S. Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: results from a national population-based study of childhood cardiomyopathy. Circulation. 2013;128:2039–46. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
