

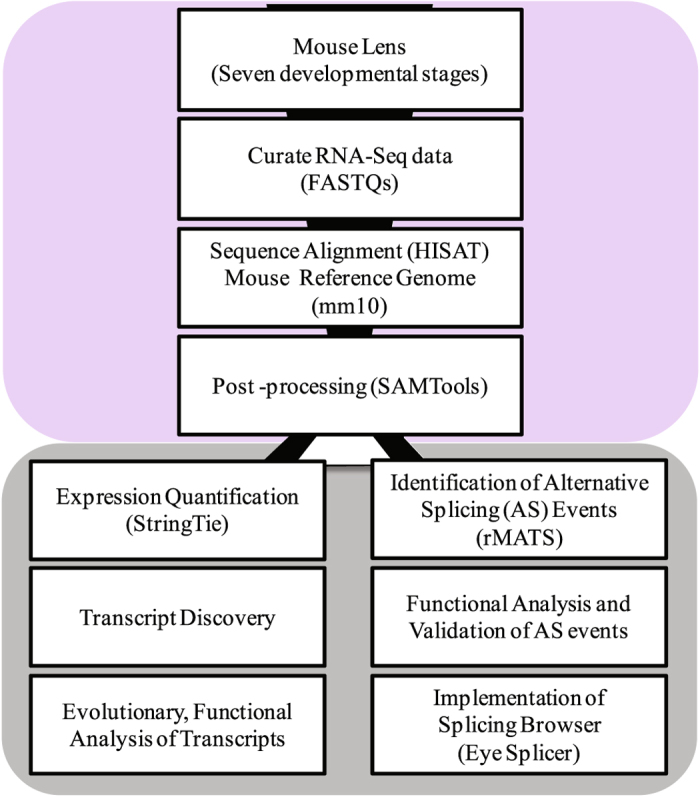
Transcriptome analysis of developing lens reveals abundance of novel transcripts and extensive splicing alterations
- PMID: 28912564
- PMCID: PMC5599659
- DOI: 10.1038/s41598-017-10615-4
Transcriptome analysis of developing lens reveals abundance of novel transcripts and extensive splicing alterations
Abstract
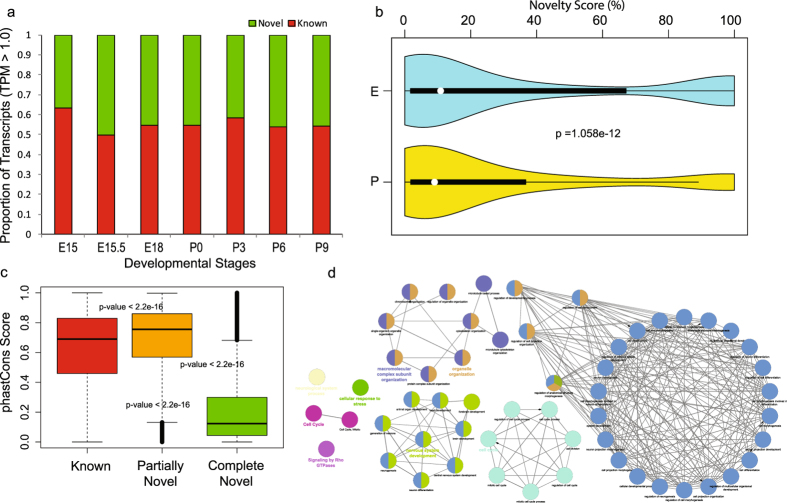
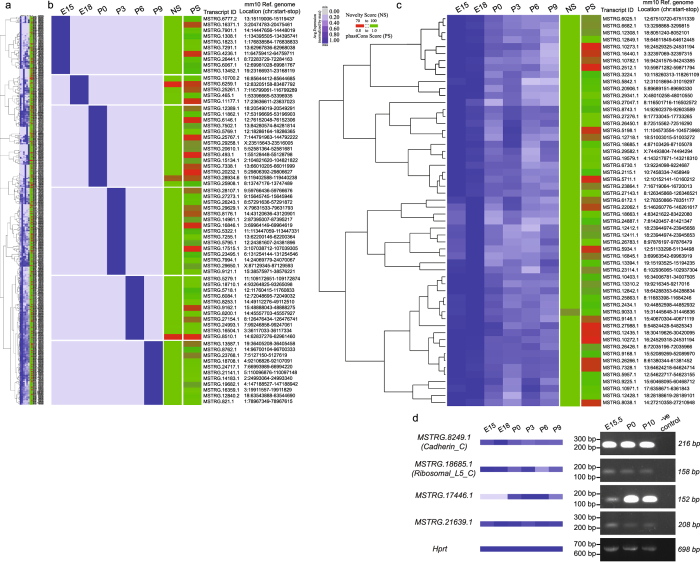
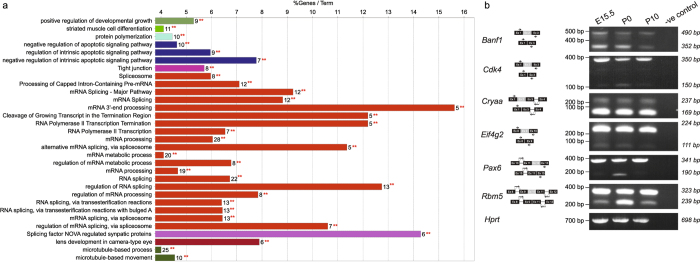
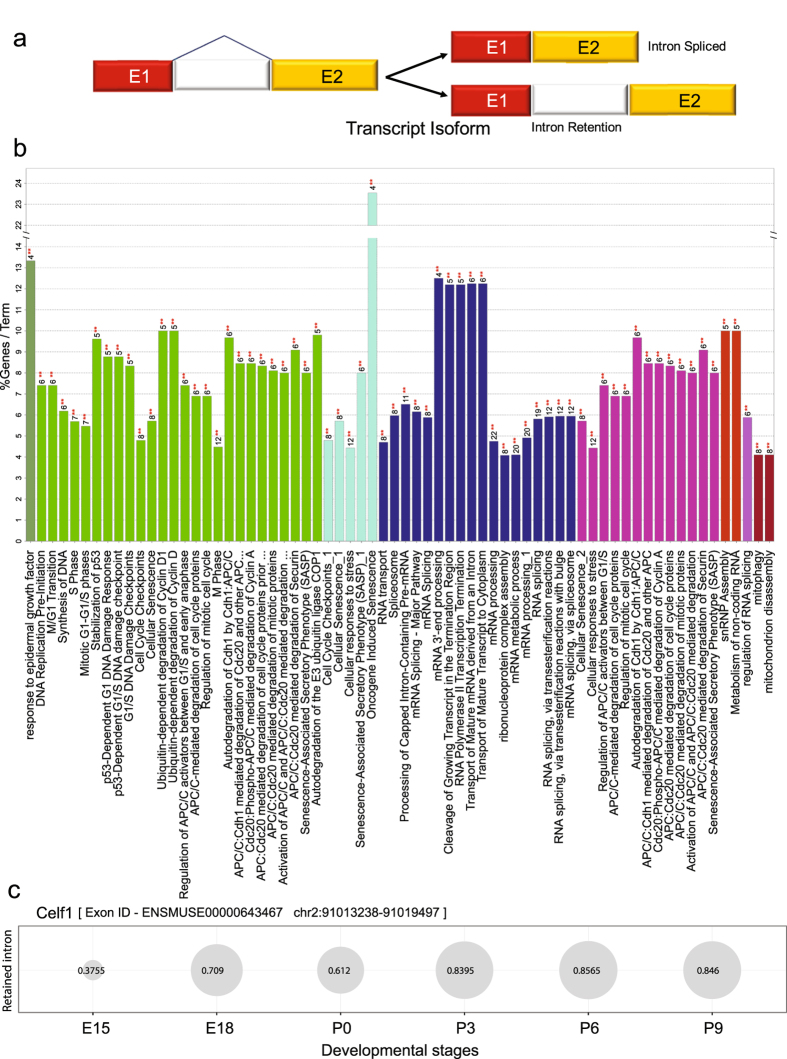
Lens development involves a complex and highly orchestrated regulatory program. Here, we investigate the transcriptomic alterations and splicing events during mouse lens formation using RNA-seq data from multiple developmental stages, and construct a molecular portrait of known and novel transcripts. We show that the extent of novelty of expressed transcripts decreases significantly in post-natal lens compared to embryonic stages. Characterization of novel transcripts into partially novel transcripts (PNTs) and completely novel transcripts (CNTs) (novelty score ≥ 70%) revealed that the PNTs are both highly conserved across vertebrates and highly expressed across multiple stages. Functional analysis of PNTs revealed their widespread role in lens developmental processes while hundreds of CNTs were found to be widely expressed and predicted to encode for proteins. We verified the expression of four CNTs across stages. Examination of splice isoforms revealed skipped exon and retained intron to be the most abundant alternative splicing events during lens development. We validated by RT-PCR and Sanger sequencing, the predicted splice isoforms of several genes Banf1, Cdk4, Cryaa, Eif4g2, Pax6, and Rbm5. Finally, we present a splicing browser Eye Splicer ( http://www.iupui.edu/~sysbio/eye-splicer/ ), to facilitate exploration of developmentally altered splicing events and to improve understanding of post-transcriptional regulatory networks during mouse lens development.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
