

A novel mechanism for Ca2+/calmodulin-dependent protein kinase II targeting to L-type Ca2+ channels that initiates long-range signaling to the nucleus
- PMID: 28916724
- PMCID: PMC5655510
- DOI: 10.1074/jbc.M117.788331
A novel mechanism for Ca2+/calmodulin-dependent protein kinase II targeting to L-type Ca2+ channels that initiates long-range signaling to the nucleus
Abstract
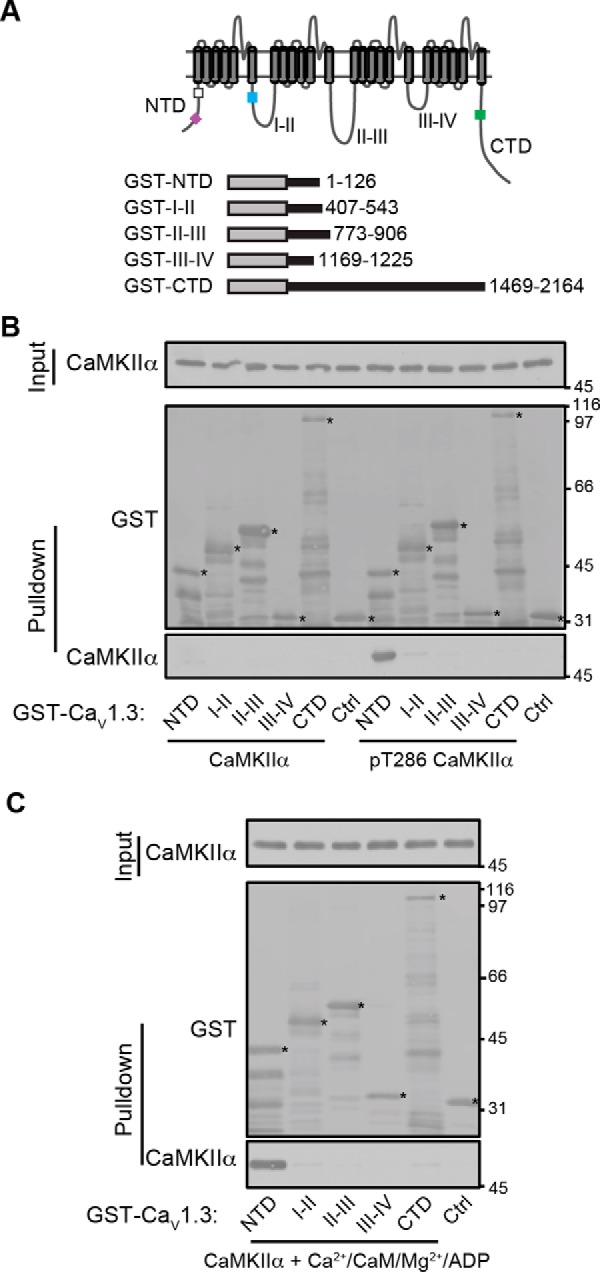
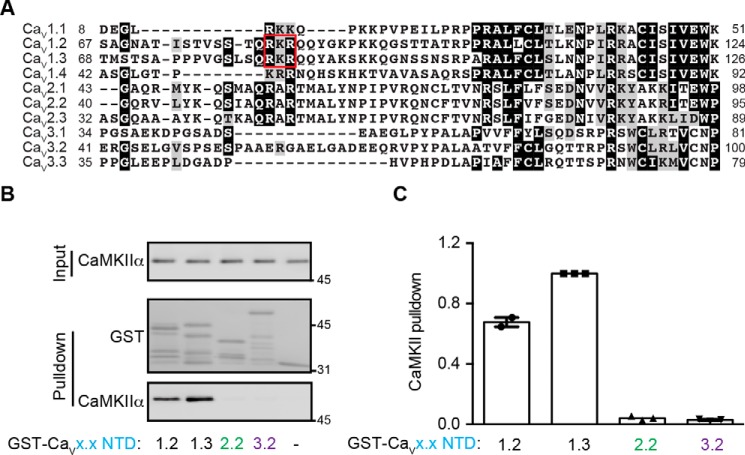
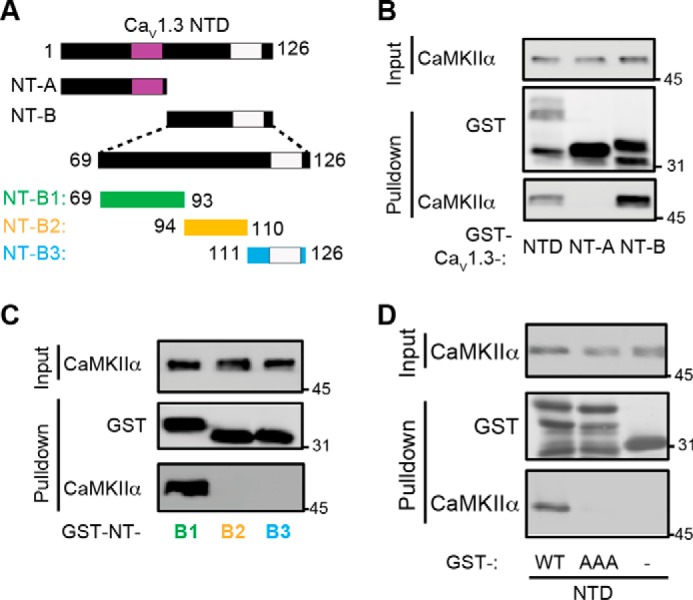
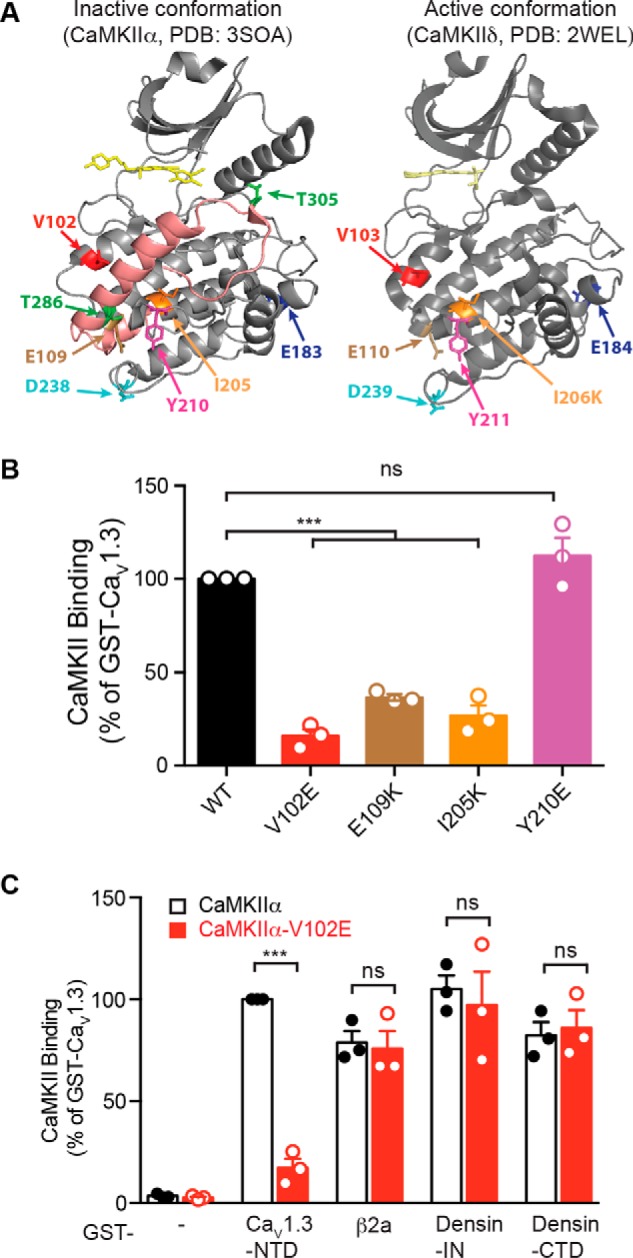
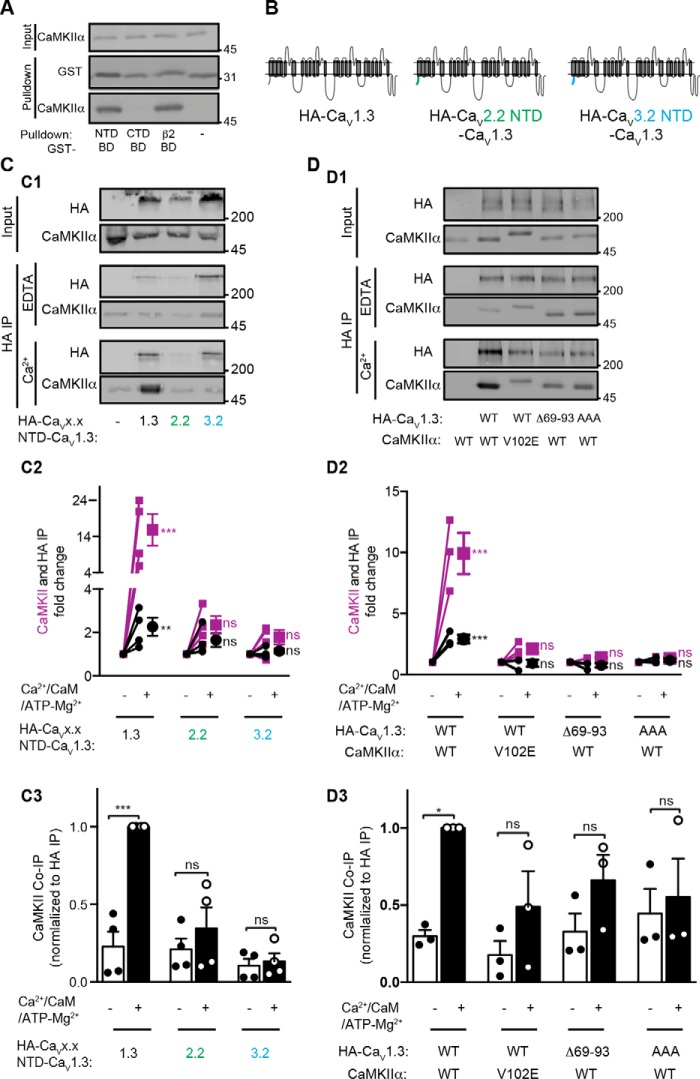
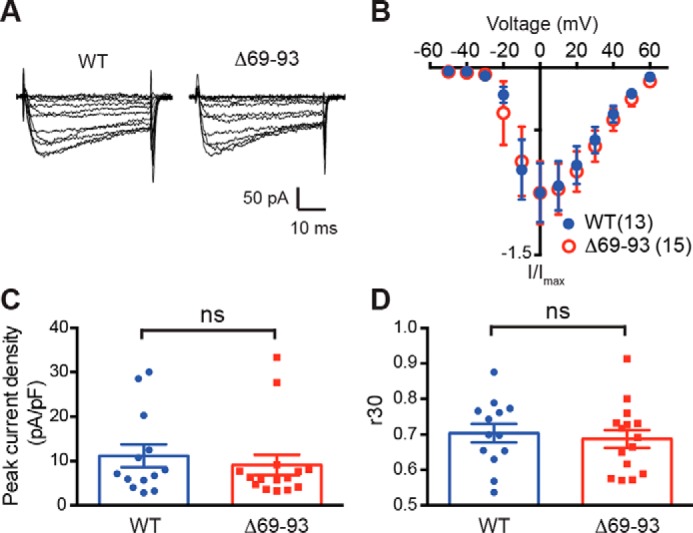
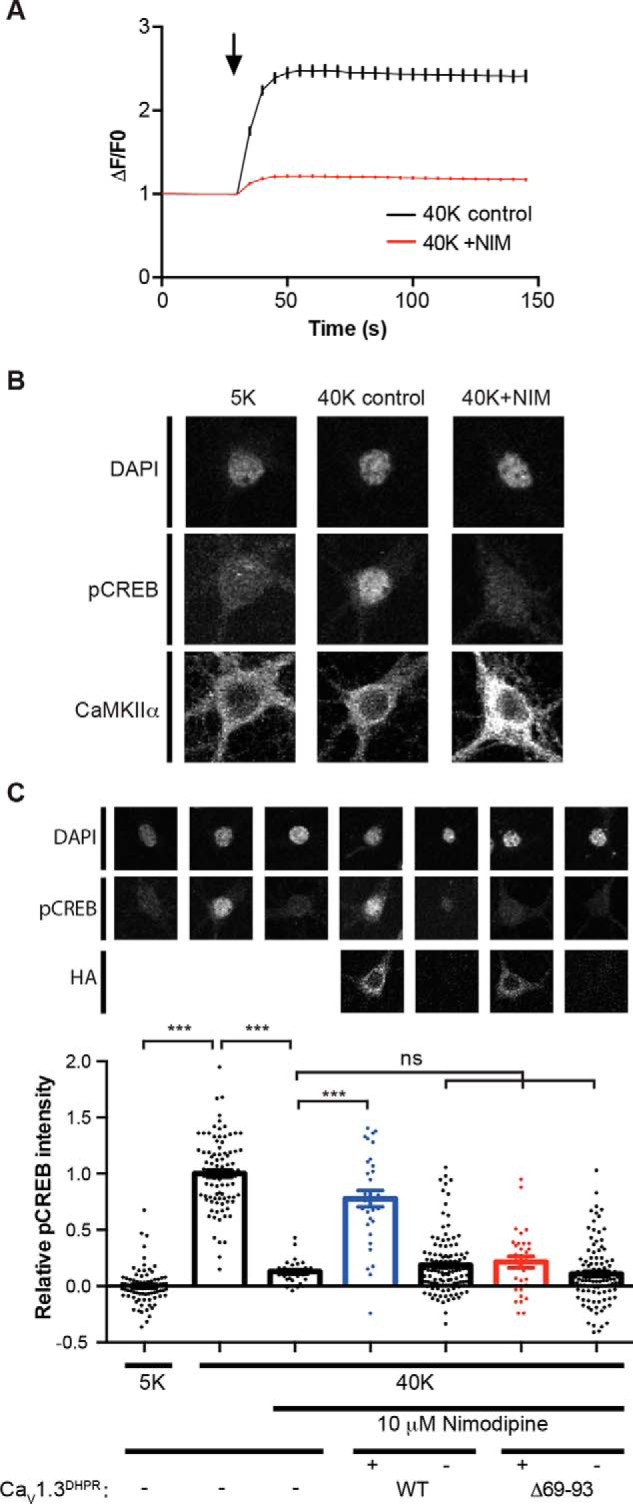
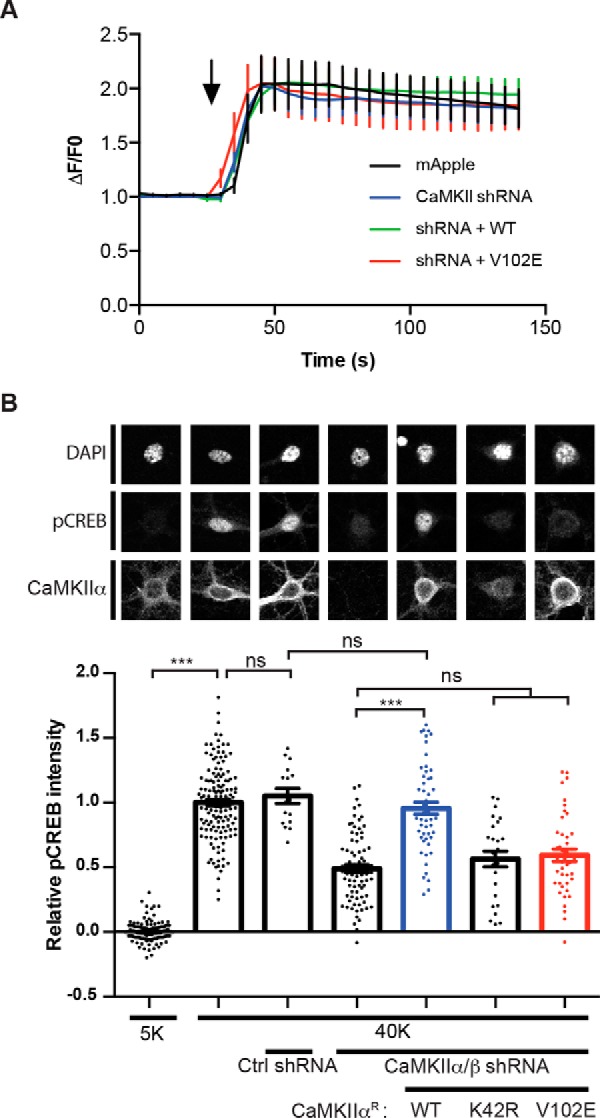
Neuronal excitation can induce new mRNA transcription, a phenomenon called excitation-transcription (E-T) coupling. Among several pathways implicated in E-T coupling, activation of voltage-gated L-type Ca2+ channels (LTCCs) in the plasma membrane can initiate a signaling pathway that ultimately increases nuclear CREB phosphorylation and, in most cases, expression of immediate early genes. Initiation of this long-range pathway has been shown to require recruitment of Ca2+-sensitive enzymes to a nanodomain in the immediate vicinity of the LTCC by an unknown mechanism. Here, we show that activated Ca2+/calmodulin-dependent protein kinase II (CaMKII) strongly interacts with a novel binding motif in the N-terminal domain of CaV1 LTCC α1 subunits that is not conserved in CaV2 or CaV3 voltage-gated Ca2+ channel subunits. Mutations in the CaV1.3 α1 subunit N-terminal domain or in the CaMKII catalytic domain that largely prevent the in vitro interaction also disrupt CaMKII association with intact LTCC complexes isolated by immunoprecipitation. Furthermore, these same mutations interfere with E-T coupling in cultured hippocampal neurons. Taken together, our findings define a novel molecular interaction with the neuronal LTCC that is required for the initiation of a long-range signal to the nucleus that is critical for learning and memory.
Keywords: Ca2+/calmodulin-dependent protein kinase II (CaMKII); cAMP-response element-binding protein (CREB); calcium channel; neuron; protein-protein interaction.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
Activity dependent Clustering of Neuronal L-Type Calcium Channels by CaMKII.bioRxiv [Preprint]. 2025 Jan 8:2025.01.08.631979. doi: 10.1101/2025.01.08.631979. bioRxiv. 2025. PMID: 39829809 Free PMC article. Preprint.
-
Neuronal L-Type Calcium Channel Signaling to the Nucleus Requires a Novel CaMKIIα-Shank3 Interaction.J Neurosci. 2020 Mar 4;40(10):2000-2014. doi: 10.1523/JNEUROSCI.0893-19.2020. Epub 2020 Feb 4. J Neurosci. 2020. PMID: 32019829 Free PMC article.
-
Clustering of CaV 1.3 L-type calcium channels by Shank3.J Neurochem. 2023 Oct;167(1):16-37. doi: 10.1111/jnc.15880. Epub 2023 Jun 30. J Neurochem. 2023. PMID: 37392026 Free PMC article.
-
Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain.Int J Mol Sci. 2017 Dec 22;19(1):20. doi: 10.3390/ijms19010020. Int J Mol Sci. 2017. PMID: 29271887 Free PMC article. Review.
-
Exploring the dominant role of Cav1 channels in signalling to the nucleus.Biosci Rep. 2012 Dec 20;33(1):97-101. doi: 10.1042/BSR20120099. Biosci Rep. 2012. PMID: 23088728 Free PMC article. Review.
Cited by
-
Genetic silencing of striatal CaV1.3 prevents and ameliorates levodopa dyskinesia.Mov Disord. 2019 May;34(5):697-707. doi: 10.1002/mds.27695. Epub 2019 Apr 19. Mov Disord. 2019. PMID: 31002755 Free PMC article.
-
Synapse-to-Nucleus ERK→CREB Transcriptional Signaling Requires Dendrite-to-Soma Ca2+ Propagation Mediated by L-Type Voltage-Gated Ca2+ Channels.J Neurosci. 2025 Jan 22;45(4):e1216242024. doi: 10.1523/JNEUROSCI.1216-24.2024. J Neurosci. 2025. PMID: 39562039 Free PMC article.
-
Role of Ca2+/Calmodulin-Dependent Protein Kinase Type II in Mediating Function and Dysfunction at Glutamatergic Synapses.Front Mol Neurosci. 2022 Jun 20;15:855752. doi: 10.3389/fnmol.2022.855752. eCollection 2022. Front Mol Neurosci. 2022. PMID: 35795689 Free PMC article. Review.
-
A revised view of the role of CaMKII in learning and memory.Nat Neurosci. 2025 Jan;28(1):24-34. doi: 10.1038/s41593-024-01809-x. Epub 2024 Nov 18. Nat Neurosci. 2025. PMID: 39558039 Review.
-
Excitation-transcription coupling, neuronal gene expression and synaptic plasticity.Nat Rev Neurosci. 2023 Nov;24(11):672-692. doi: 10.1038/s41583-023-00742-5. Epub 2023 Sep 29. Nat Rev Neurosci. 2023. PMID: 37773070 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
- R01 DK097392/DK/NIDDK NIH HHS/United States
- R01 NS078291/NS/NINDS NIH HHS/United States
- P60 DK020593/DK/NIDDK NIH HHS/United States
- R01 HD061543/HD/NICHD NIH HHS/United States
- P30 DK058404/DK/NIDDK NIH HHS/United States
- U2C DK059637/DK/NIDDK NIH HHS/United States
- P30 EY008126/EY/NEI NIH HHS/United States
- R01 MH063232/MH/NIMH NIH HHS/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- F31 MH109196/MH/NIMH NIH HHS/United States
- U24 DK059637/DK/NIDDK NIH HHS/United States
- U54 HD083211/HD/NICHD NIH HHS/United States
- R01 DC009433/DC/NIDCD NIH HHS/United States
- R01 NS084190/NS/NINDS NIH HHS/United States
- P30 DK020593/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
