

Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-β Pathway
- PMID: 28918914
- PMCID: PMC6192529
- DOI: 10.1053/j.gastro.2017.09.007
Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-β Pathway
Abstract
Background & aims: Development of hepatocellular carcinoma (HCC) is associated with alterations in the transforming growth factor-beta (TGF-β) signaling pathway, which regulates liver inflammation and can have tumor suppressor or promoter activities. Little is known about the roles of specific members of this pathway at specific of HCC development. We took an integrated approach to identify and validate the effects of changes in this pathway in HCC and identify therapeutic targets.
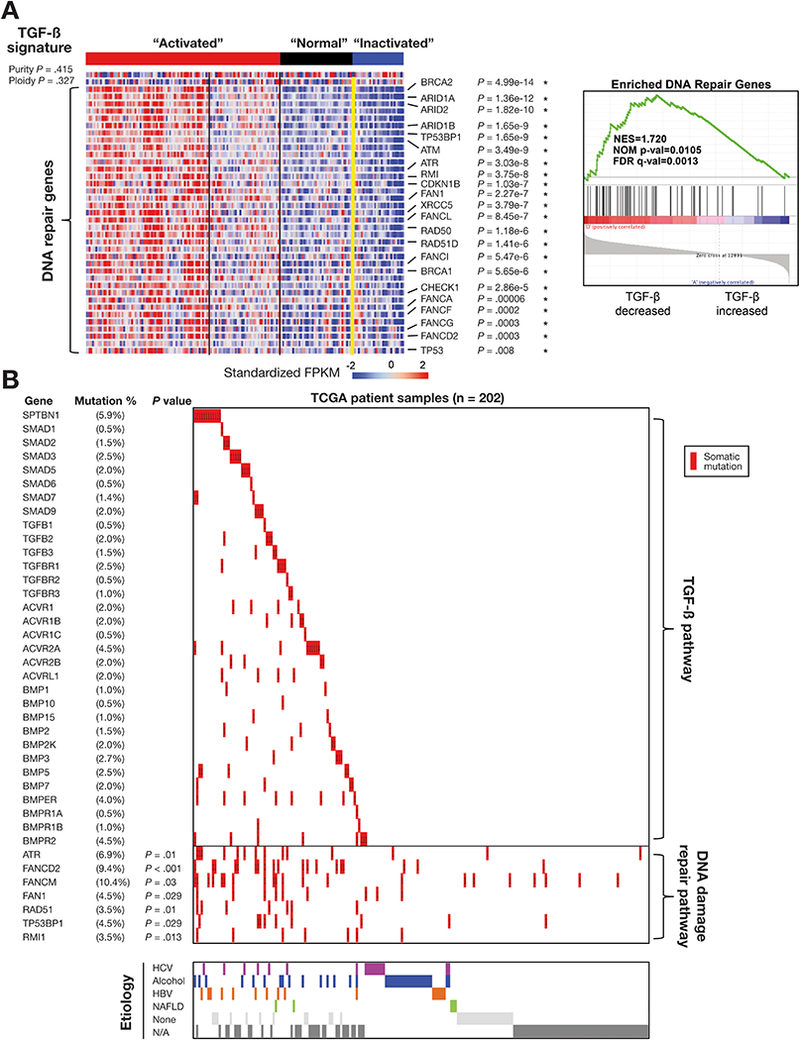
Methods: We performed transcriptome analyses for a total of 488 HCCs that include data from The Cancer Genome Atlas. We also screened 301 HCCs reported in the Catalogue of Somatic Mutations in Cancer and 202 from Cancer Genome Atlas for mutations in genome sequences. We expressed mutant forms of spectrin beta, non-erythrocytic 1 (SPTBN1) in HepG2, SNU398, and SNU475 cells and measured phosphorylation, nuclear translocation, and transcriptional activity of SMAD family member 3 (SMAD3).
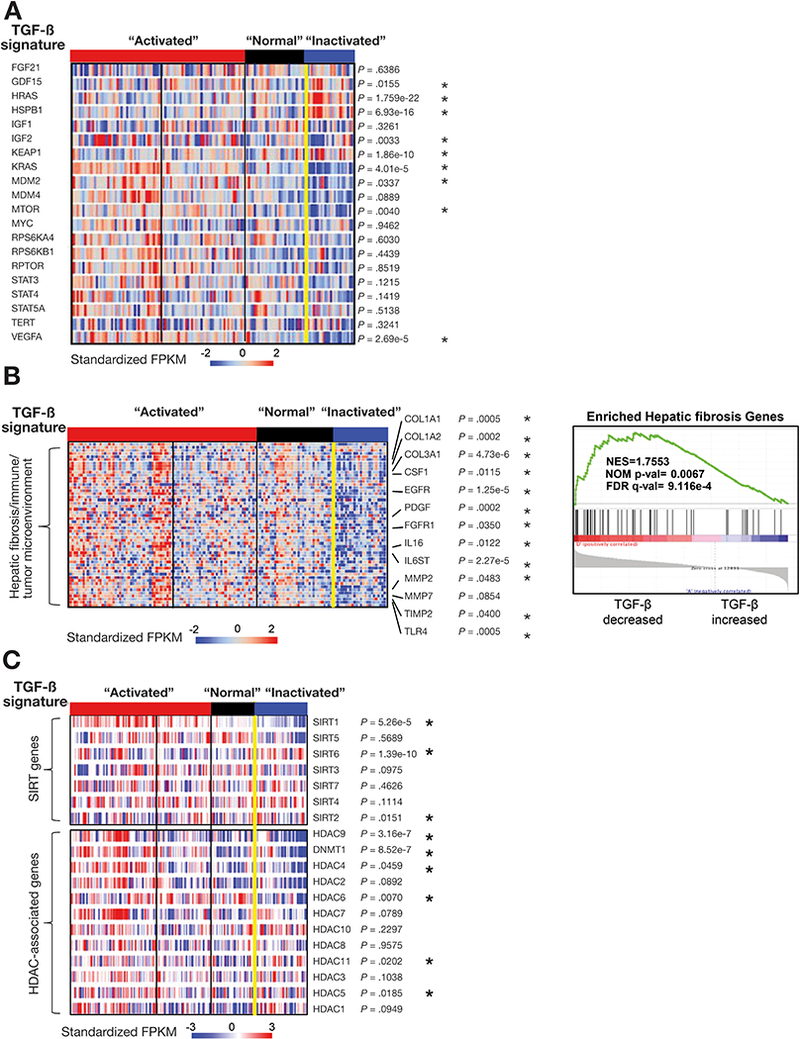
Results: We found somatic mutations in at least 1 gene whose product is a member of TGF-β signaling pathway in 38% of HCC samples. SPTBN1 was mutated in the largest proportion of samples (12 of 202, 6%). Unsupervised clustering of transcriptome data identified a group of HCCs with activation of the TGF-β signaling pathway (increased transcription of genes in the pathway) and a group of HCCs with inactivation of TGF-β signaling (reduced expression of genes in this pathway). Patients with tumors with inactivation of TGF-β signaling had shorter survival times than patients with tumors with activation of TGF-β signaling (P = .0129). Patterns of TGF-β signaling correlated with activation of the DNA damage response and sirtuin signaling pathways. HepG2, SNU398, and SNU475 cells that expressed the D1089Y mutant or with knockdown of SPTBN1 had increased sensitivity to DNA crosslinking agents and reduced survival compared with cells that expressed normal SPTBN1 (controls).
Conclusions: In genome and transcriptome analyses of HCC samples, we found mutations in genes in the TGF-β signaling pathway in almost 40% of samples. These correlated with changes in expression of genes in the pathways; up-regulation of genes in this pathway would contribute to inflammation and fibrosis, whereas down-regulation would indicate loss of TGF-β tumor suppressor activity. Our findings indicate that therapeutic agents for HCCs can be effective, based on genetic features of the TGF-β pathway; agents that block TGF-β should be used only in patients with specific types of HCCs.
Keywords: COSMIC; Gene Regulation; Genetics; Immune Response; Liver Cancer.
Copyright © 2018 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest
None
Figures




Comment in
-
The Role of Transforming Growth Factor-β in Human Hepatocarcinogenesis: Mechanistic and Therapeutic Implications From an Integrative Multiomics Approach.Gastroenterology. 2018 Jan;154(1):17-20. doi: 10.1053/j.gastro.2017.11.015. Epub 2017 Nov 21. Gastroenterology. 2018. PMID: 29174548 No abstract available.
References
-
- GLOBOCAN 2012: Liver cancer: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. International Agency for Research on Cancer: World Health Organization. 2017. (Accessed on Jun 11, 2017 at http://globocan.iarc.fr/old/FactSheets/cancers/liver-new.asp).
-
- Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016;150:835–53. - PubMed
-
- Totoki Y, Tatsuno K, Covington KR, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 2014;46:1267–73. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
