

Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma
- PMID: 28923912
- PMCID: PMC6456057
- DOI: 10.1158/2159-8290.CD-17-0682
Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma
Expression of concern in
-
Expression of Concern: Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma.Cancer Discov. 2024 Jul 1;14(7):1356. doi: 10.1158/2159-8290.CD-24-0612. Cancer Discov. 2024. PMID: 38946327 No abstract available.
Abstract
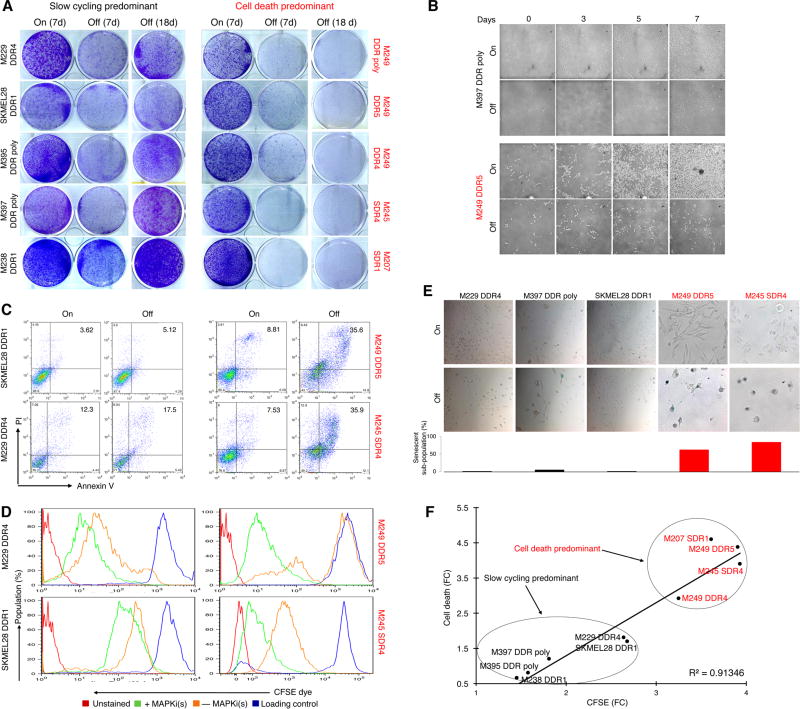
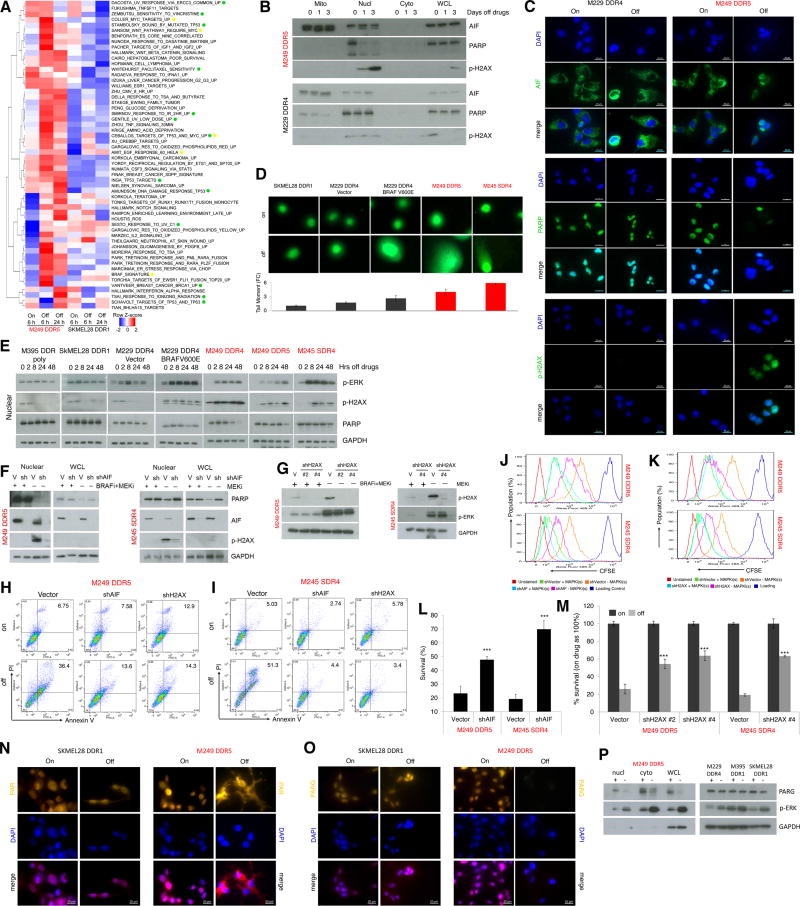
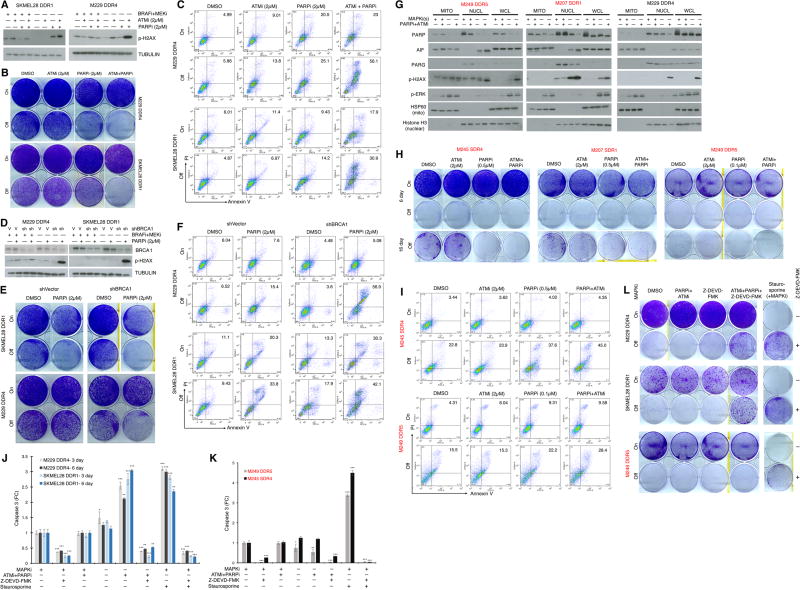
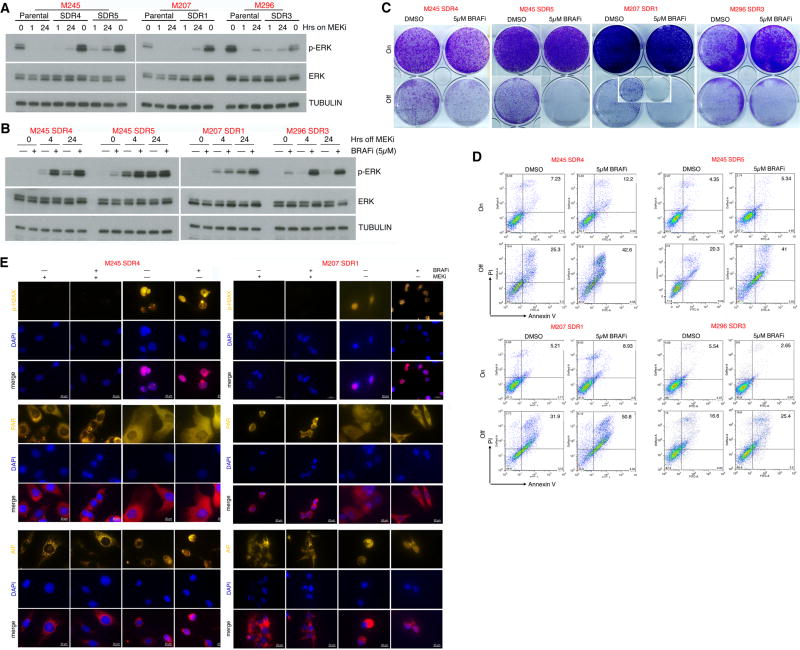
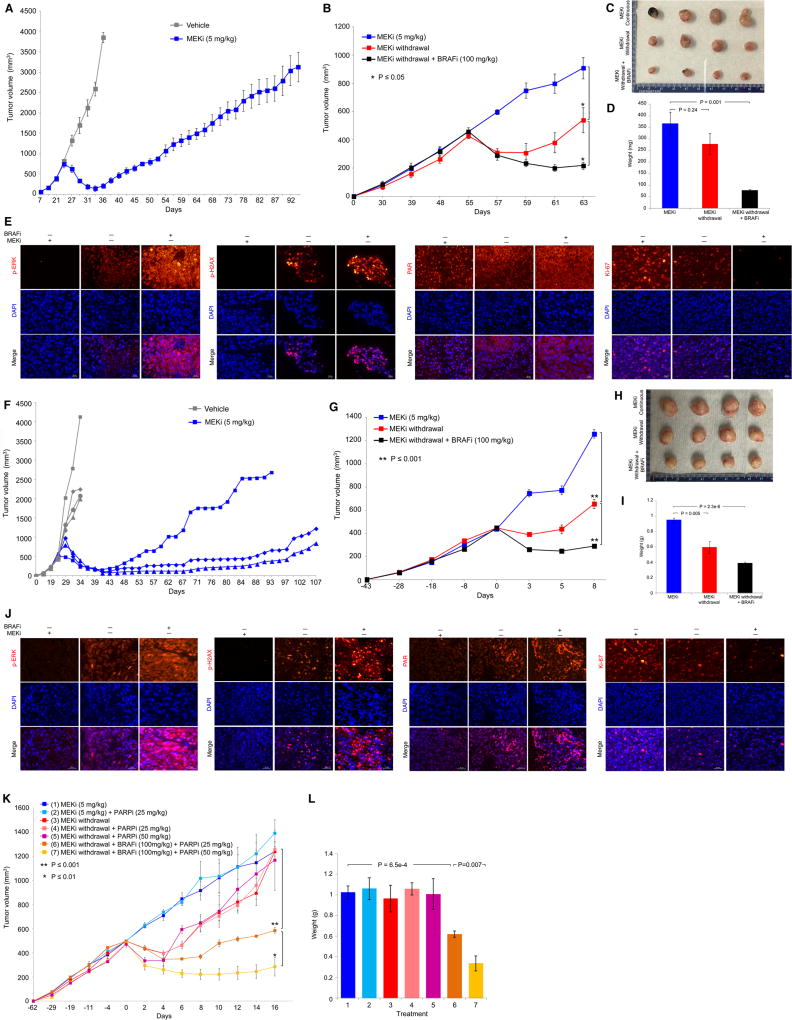
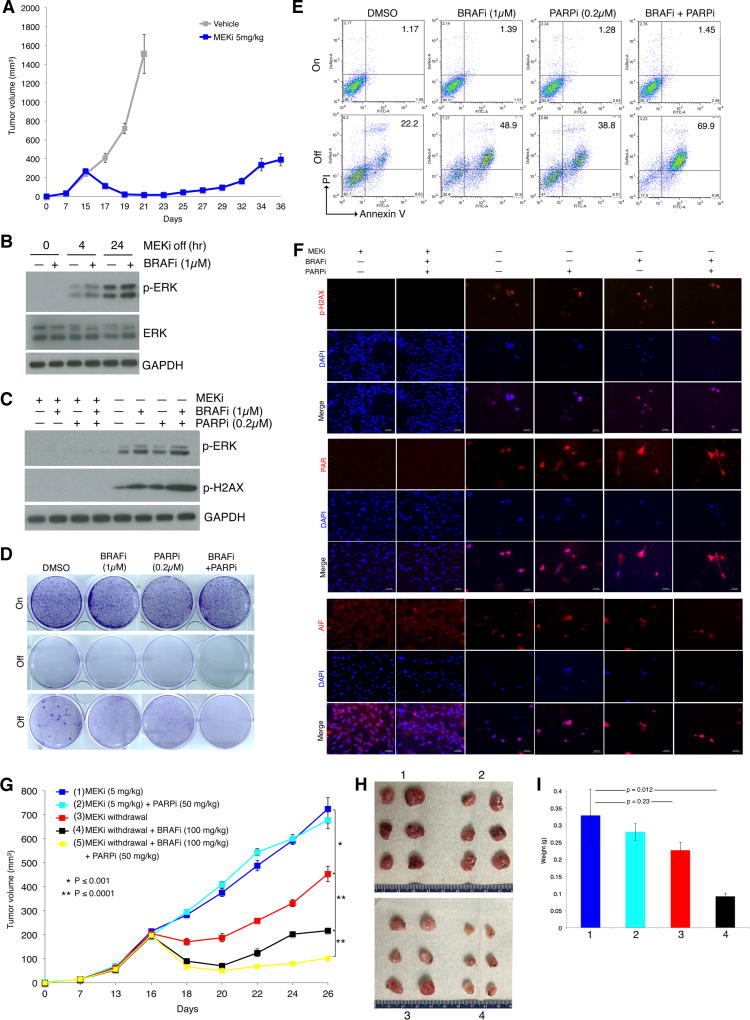
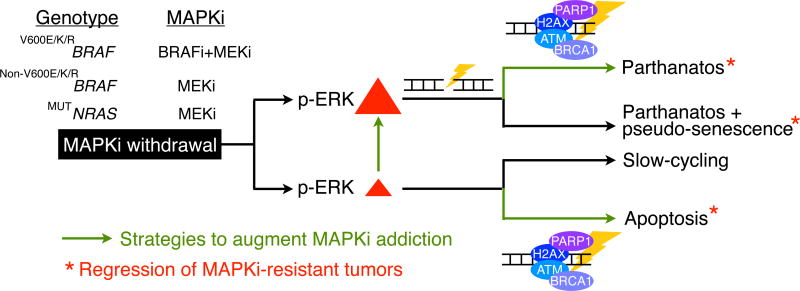
Melanoma resistant to MAPK inhibitors (MAPKi) displays loss of fitness upon experimental MAPKi withdrawal and, clinically, may be resensitized to MAPKi therapy after a drug holiday. Here, we uncovered and therapeutically exploited the mechanisms of MAPKi addiction in MAPKi-resistant BRAFMUT or NRASMUT melanoma. MAPKi-addiction phenotypes evident upon drug withdrawal spanned transient cell-cycle slowdown to cell-death responses, the latter of which required a robust phosphorylated ERK (pERK) rebound. Generally, drug withdrawal-induced pERK rebound upregulated p38-FRA1-JUNB-CDKN1A and downregulated proliferation, but only a robust pERK rebound resulted in DNA damage and parthanatos-related cell death. Importantly, pharmacologically impairing DNA damage repair during MAPKi withdrawal augmented MAPKi addiction across the board by converting a cell-cycle deceleration to a caspase-dependent cell-death response or by furthering parthanatos-related cell death. Specifically in MEKi-resistant NRASMUT or atypical BRAFMUT melanoma, treatment with a type I RAF inhibitor intensified pERK rebound elicited by MEKi withdrawal, thereby promoting a cell death-predominant MAPKi-addiction phenotype. Thus, MAPKi discontinuation upon disease progression should be coupled with specific strategies that augment MAPKi addiction.Significance: Discontinuing targeted therapy may select against drug-resistant tumor clones, but drug-addiction mechanisms are ill-defined. Using melanoma resistant to but withdrawn from MAPKi, we defined a synthetic lethality between supraphysiologic levels of pERK and DNA damage. Actively promoting this synthetic lethality could rationalize sequential/rotational regimens that address evolving vulnerabilities. Cancer Discov; 8(1); 74-93. ©2017 AACR.See related commentary by Stern, p. 20This article is highlighted in the In This Issue feature, p. 1.
©2017 American Association for Cancer Research.
Conflict of interest statement
Authors declare no conflicts of interest.
Figures
Comment in
-
Keeping Tumors Out of the MAPK Fitness Zone.Cancer Discov. 2018 Jan;8(1):20-23. doi: 10.1158/2159-8290.CD-17-1243. Cancer Discov. 2018. PMID: 29311225
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
