

Emerging Concepts in TCR Specificity: Rationalizing and (Maybe) Predicting Outcomes
- PMID: 28923982
- PMCID: PMC5679125
- DOI: 10.4049/jimmunol.1700744
Emerging Concepts in TCR Specificity: Rationalizing and (Maybe) Predicting Outcomes
Abstract
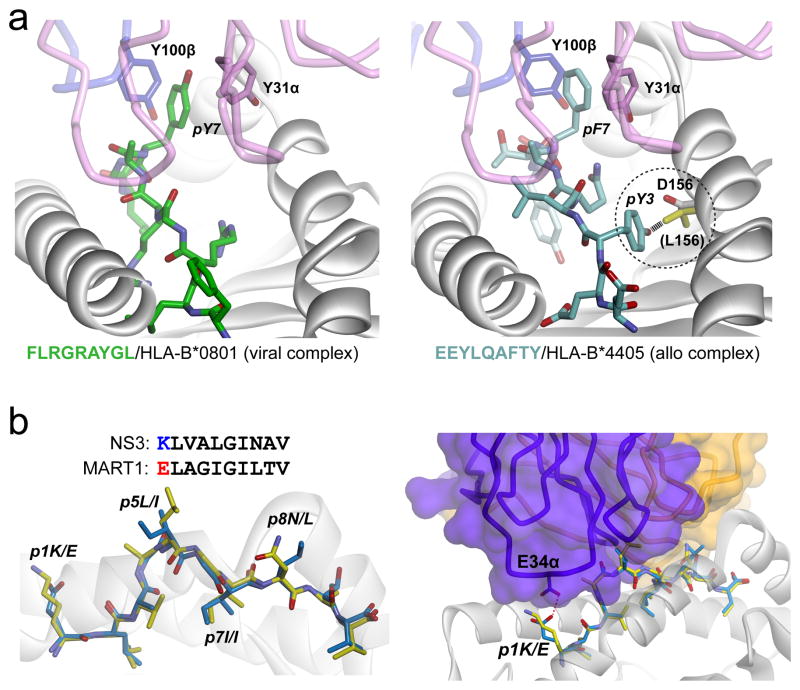
T cell specificity emerges from a myriad of processes, ranging from the biological pathways that control T cell signaling to the structural and physical mechanisms that influence how TCRs bind peptides and MHC proteins. Of these processes, the binding specificity of the TCR is a key component. However, TCR specificity is enigmatic: TCRs are at once specific but also cross-reactive. Although long appreciated, this duality continues to puzzle immunologists and has implications for the development of TCR-based therapeutics. In this review, we discuss TCR specificity, emphasizing results that have emerged from structural and physical studies of TCR binding. We show how the TCR specificity/cross-reactivity duality can be rationalized from structural and biophysical principles. There is excellent agreement between predictions from these principles and classic predictions about the scope of TCR cross-reactivity. We demonstrate how these same principles can also explain amino acid preferences in immunogenic epitopes and highlight opportunities for structural considerations in predictive immunology.
Copyright © 2017 by The American Association of Immunologists, Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
